The radial function of basis orbitals can be variationally optimized using the orbital optimization method [28]. As an illustration of the orbital optimization, let us explain it using a methane molecule of which input file is 'Methane_OO.dat'. In the orbital optimization method the optimized orbitals are expressed by the linear combination of primitive orbitals, and obtained by variationally optimizing the contraction coefficients. The number of the primitive and optimized orbitals in the optimization are specified by
<Definition.of.Atomic.Species
H H5.0-s4>1 H_CA13
C C5.0-s4>1p4>1 C_CA13
Definition.of.Atomic.Species>
For 'H' one optimized radial function for the s-orbital is obtained from
the linear combination of four primitive radial functions.
Similarly, one optimized radial function for the s-(p-)orbital is obtained from
the linear combination of four primitive radial functions for 'C'.
In addition, the following keywords are set in the input file as follows:
orbitalOpt.Method species # Off|Species|Atoms
orbitalOpt.Opt.Method EF # DIIS|EF
orbitalOpt.SD.step 0.001 # default=0.001
orbitalOpt.HistoryPulay 30 # default=15
orbitalOpt.StartPulay 10 # default=1
orbitalOpt.scf.maxIter 60 # default=40
orbitalOpt.Opt.maxIter 140 # default=100
orbitalOpt.per.MDIter 20 # default=1000000
orbitalOpt.criterion 1.0e-4 # default=1.0e-4
CntOrb.fileout on # on|off, default=off
Num.CntOrb.Atoms 2 # default=1
<Atoms.Cont.Orbitals
1
2
Atoms.Cont.Orbitals>
Then, we execute OpenMX as:
% ./openmx Methane_OO.dat
When the execution is completed normally, you can find the history
of orbital optimization in the file 'met_oo.out' as:
***********************************************************
***********************************************************
History of orbital optimization MD= 1
********* Gradient Norm ((Hartree/borh)^2) ********
Required criterion= 0.000100000000
***********************************************************
iter= 1 Gradient Norm= 0.057098961101 Uele= -3.217161102876
iter= 2 Gradient Norm= 0.044668461503 Uele= -3.220120116009
iter= 3 Gradient Norm= 0.034308306321 Uele= -3.223123238394
iter= 4 Gradient Norm= 0.025847573248 Uele= -3.226177980300
iter= 5 Gradient Norm= 0.019106400842 Uele= -3.229294858054
iter= 6 Gradient Norm= 0.013893824906 Uele= -3.232489198284
iter= 7 Gradient Norm= 0.010499500005 Uele= -3.235304178159
iter= 8 Gradient Norm= 0.008362635043 Uele= -3.237652870812
iter= 9 Gradient Norm= 0.006959703539 Uele= -3.239618540761
iter= 10 Gradient Norm= 0.005994816379 Uele= -3.241268535418
iter= 11 Gradient Norm= 0.005298095979 Uele= -3.242657118263
iter= 12 Gradient Norm= 0.003059655878 Uele= -3.250892948269
iter= 13 Gradient Norm= 0.001390201488 Uele= -3.255123241210
iter= 14 Gradient Norm= 0.000780925380 Uele= -3.255179362845
iter= 15 Gradient Norm= 0.000726631072 Uele= -3.255263012792
iter= 16 Gradient Norm= 0.000390930576 Uele= -3.250873416989
iter= 17 Gradient Norm= 0.000280785975 Uele= -3.250333677139
iter= 18 Gradient Norm= 0.000200668585 Uele= -3.252345643243
iter= 19 Gradient Norm= 0.000240367596 Uele= -3.254238199726
iter= 20 Gradient Norm= 0.000081974594 Uele= -3.258146794679
In most cases, 20-50 iterative steps are enough to achieve
a sufficient convergence. The comparison between the primitive basis
orbitals and the optimized orbitals in the total energy
is given by
Primitive basis orbitals
Utot = -7.992569945749 (Hartree)
Optimized orbitals by the orbital optimization
Utot = -8.133746986502 (Hartree)
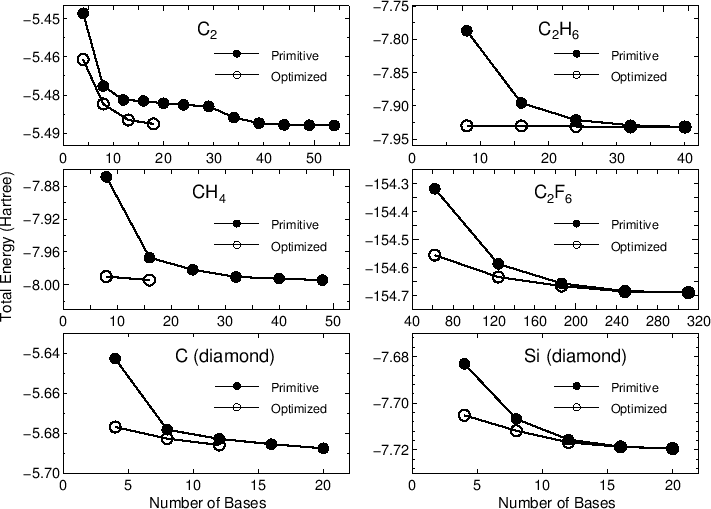 |
The following two options are available for the keyword 'orbitalOpt.Method': 'atoms' in which basis obitals on each atom are fully optimized, 'species' in which basis obitals on each species are optimized.
The radial functions of basis orbitals are optimized
with a constraint that the radial wave function ![]() is independent
of the magnetic quantum number, which guarantees the rotational invariance
of the total energy. However, the optimized orbital on all the atoms can
be different from eath other.
is independent
of the magnetic quantum number, which guarantees the rotational invariance
of the total energy. However, the optimized orbital on all the atoms can
be different from eath other.
Basis orbitals in atoms with the same species name, that you define in 'Definition.of.Atomic.Species', are optimized as the same orbitals. If you want to assign the same orbitals to atoms with almost the same chemical environment, and optimize these orbitals, this scheme could be quite convenient. As well as 'atoms', the optimized radial functions are independent of the magnetic quantum number, which guarantees the rotational invariance of the total energy.
Although the same information is available in the section 'Input file',
for convenience the details of the other keywords are listed below:
orbitalOpt.scf.maxIter
The maximum number of SCF iterations in the orbital optimization is
specified by the keyword 'orbitalOpt.scf.maxIter'.
orbitalOpt.Opt.maxIter
The maximum number of iterations for the orbital optimization is specified
by the keyword 'orbitalOpt.Opt.maxIter'. The iteration loop for the orbital
optimization is terminated at the number specified by 'orbitalOpt.Opt.maxIter'
even when a convergence criterion is not satisfied.
orbitalOpt.Opt.Method
Two schemes for the optimization of orbitals are available:
'EF' which is an eigenvector following method, 'DIIS' which is
the direct inversion method in iterative subspace.
The algorithms are basically same as for the geometry optimization.
Either 'EF' or 'DIIS' is chosen by the keyword, 'orbitalOpt.Opt.Method'.
orbitalOpt.StartPulay
The quasi Newton method, 'EF' and 'DIIS' starts from the optimization step
specified by the keyword 'orbitalOpt.StartPulay'.
orbitalOpt.HistoryPulay
The keyword 'orbitalOpt.HistoryPulay' specifies the number of previous steps
to estimate the next input contraction coefficients used in the quasi Newton
method, 'EF' and 'DIIS'.
orbitalOpt.SD.step
Steps before moving the quasi Newton method, 'EF' and 'DIIS' is performed
by the steepest decent method. The prefactor used in the steepest decent
method is specified by the keyword 'orbitalOpt.SD.step'. In most cases,
orbitalOpt.SD.step of 0.001 can be a good prefactor.
orbitalOpt.criterion
The keyword 'orbitalOpt.criterion' specifies a convergence criterion
((Hartree/borh)![]() ) for the orbital optimization. The iterations loop is
finished when a condition, Norm of derivatives
) for the orbital optimization. The iterations loop is
finished when a condition, Norm of derivatives![]() orbitalOpt.criterion,
is satisfied.
orbitalOpt.criterion,
is satisfied.
CntOrb.fileout
If you want to output the optimized radial orbitals to files,
then the keyword 'CntOrb.fileout' must be ON.
Num.CntOrb.Atoms
The keyword 'Num.CntOrb.Atoms' gives the number of atoms whose
optimized radial orbitals are output to files.
Atoms.Cont.Orbitals
The keyword 'Atoms.Cont.Orbitals' specifies the atom number,
which was given by the first column in the specification of
the keyword 'Atoms.SpeciesAndCoordinates' for the output
of optimized orbitals as follows:
<Atoms.Cont.Orbitals
1
2
Atoms.Cont.Orbitals>
The beginning of the description must be '