To calculate an electronic structure with an arbitrary spin orientation in the non-collinear DFT, OpenMX Ver. 3.7 provides a constraint functional which gives a penalty unless the difference between the calculated spin orientation and the initial one is zero [11]. The constraint DFT for the non-collinear spin orientation is available by the following keywords:
scf.Constraint.NC.Spin on # on|off, default=off
scf.Constraint.NC.Spin.v 0.5 # default=0.0(eV)
You can switch on the keyword
'scf.Constraint.NC.Spin' and give
a magnitude by 'scf.Constraint.NC.Spin.v'
which determines the
strength of constraint, when the constraint for the spin orientation
is introduced. The constraint is applied on each atom
by specifying a switch as follows:
<Atoms.SpeciesAndCoordinates
1 Cr 0.00000 0.00000 0.00000 7.0 5.0 -20.0 0.0 1 off
2 Cr 0.00000 2.00000 0.00000 7.0 5.0 20.0 0.0 1 off
Atoms.SpeciesAndCoordinates>
The '1' in the 10th column means that the constraint is applied,
and '0' no constraint. The method constrains only the spin orientation.
Therefore, the magnitude of spin can vary. Also the constraint scheme
is compatible with the LDA+U calculation explained in the Section 'LDA+U'.
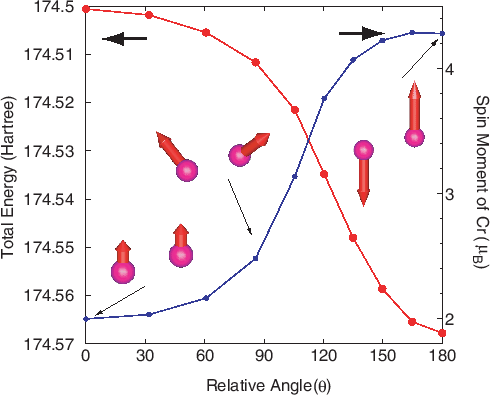
As an illustration of this method,
the dependence of the total energy and magnetic moment in a chromium
dimer on the relative angle between two local spins is shown
in Fig. 28. You can trace the calculation using an input file
'Cr2_CNC.dat' in the directory 'work'.
 |