Next: Unfolding of band structures Up: Unfolding method for band Previous: Unfolding method for band Contents Index
First, let us analyze how each band can be decomposed into each contribution of pseudo-atomic orbital in a band structure calculation for a primitive cell of SiC in a two-dimensional honeycomb structure without imperfection. Note that unfolding bands is not performed in this case. The SCF calculation for the primitive cell of the two-dimensional SiC can be performed as
% mpirun -np 16 openmx SiC_Primitive.dat > sic_primitive.std &
Species.Number 3 <Definition.of.Atomic.Species C C7.0-s2p2d1 C_PBE13 Si Si7.0-s2p2d1 Si_PBE13 Te Te11.0-s2p2d2f1 E Definition.of.Atomic.Species> Atoms.Number 3 Atoms.SpeciesAndCoordinates.Unit FRAC # Ang|AU <Atoms.SpeciesAndCoordinates 1 C 0.33333333 0.66666666 0.50000000 2.0 2.0 2 Si 0.66666666 0.33333333 0.50000000 2.0 2.0 3 Te 0.00000000 0.00000000 0.50000000 0.0 0.0 Atoms.SpeciesAndCoordinates> Atoms.UnitVectors.Unit Ang # Ang|AU <Atoms.UnitVectors 3.0690 0.0000000000 0.000 -1.5345 2.6578319641 0.000 0.0000 0.0000000000 10.000 Atoms.UnitVectors>
where an empty atom having basis functions with a long tail is allocated at the center of the hexagon
in order to improve description of conduction bands. Since the keyword 'Band.dispersion' is switched on as
Band.dispersion on # on|off, default=off Band.Nkpath 3 <Band.kpath 60 0.33333333333 0.33333333333 0.00000000000 0.00000000000 0.00000000000 0.00000000000 K G 52 0.00000000000 0.00000000000 0.00000000000 0.50000000000 0.00000000000 0.00000000000 G M 30 0.50000000000 0.00000000000 0.00000000000 0.33333333333 0.33333333333 0.00000000000 M K Band.kpath>
Keywords relevant to analysis of band structure
For the calculation, the following keywords are also given as well
Unfolding.Electronic.Band on # on|off, default=off Unfolding.LowerBound -10.0 # default=-10 eV Unfolding.UpperBound 6.0 # default= 10 eV Unfolding.Nkpoint 4 <Unfolding.kpoint K 0.33333333333 0.33333333333 0.0000000000 G 0.00000000000 0.00000000000 0.0000000000 M 0.50000000000 0.00000000000 0.0000000000 K 0.33333333333 0.33333333333 0.0000000000 Unfolding.kpoint> Unfolding.desired_totalnkpt 30
The specification of the keywords above are listed below.
To analyze and/or unfold bands the keyword should be switched on. The default is 'off'.
The keyword 'Unfolding.LowerBound' specifies the lower bound for the energy of bands in the analysis, where the energy is taken as the relative energy to the chemical potential. The default value is -10 eV.
The keyword 'Unfolding.UpperBound' specifies the upper bound for the energy of bands in the analysis, where the energy is taken as the relative energy to the chemical potential. The default value is 10 eV.
The keyword 'Unfolding.Nkpoint' specifies the number of k-points appearing in the keyword 'Unfolding.kpoint'.
The keyword 'Unfolding.kpoint' specifies the k-points of which number is given by the keyword 'Unfolding.Nkpoint'. In the above case four k-points are given, and three k-paths conecting the two k-points are considered, i.e., one from 'K' to 'G', one from 'G' to 'M', and one from 'M' to 'K'. Along the k-paths the analysis of bands is performed. Note that the unit for the k-points specified by the keyword is the reciprocal lattice vectors of the unit cell given by the keyword 'Atoms.UnitVectors' in this case, while the cell vectors can be changed to perform unfolding of bands by utilizing a keyword 'Unfolding.ReferenceVectors'. At this moment, it should be noted that the reciprocal lattice vectors of the unit cell given by the keyword 'Atoms.UnitVectors' is automatically used unless the keyword 'Unfolding.ReferenceVectors' is explicitly specified. The keyword 'Unfolding.ReferenceVectors' will be explained in the next subsection.
The k-paths specified by the keyword 'Unfolding.kpoint' are divided with a nearly equal spacing, where the spacing is estimated by (the total length of all the k-paths)/Unfolding.desired_totalnkpt. Starting from the estimated spacing, the actual spacing is automatically adjusted from one k-path to another k-path so that the k-points specified by the keyword 'Unfolding.kpoint' can be always included in the analysis.
Output files relevant to analysis of band structure
After getting the SCF convergence, the following files related to analyzing and/or unfolding bands will be generated.
The total spectral weight given by Eq. (24) in Ref. [98] is stored.
The first, second, and third columns correspond to the distance measured from the first k-point given
by the keyword 'Unfolding.kpoint' in the unit of Bohr![]() , energy relative to the chemical potential
in the unit of eV, and the total spectral weight, respectively.
In the spin-polarized calculation, 'System.Name.unfold_totdn' is also generaged for the down spin case,
while only a single file 'System.Name.unfold_tot' is generated in the non-collinear calculation.
, energy relative to the chemical potential
in the unit of eV, and the total spectral weight, respectively.
In the spin-polarized calculation, 'System.Name.unfold_totdn' is also generaged for the down spin case,
while only a single file 'System.Name.unfold_tot' is generated in the non-collinear calculation.
The orbitally decomposed spectral weights given by Eq. (26) in Ref. [98] are stored.
The first, second, and subsequent columns correspond to the distance measured from the first k-point given
by the keyword 'Unfolding.kpoint' in the unit of Bohr![]() , energy relative to the chemical potential
in the unit of eV, and the orbitally decomposed spectral weights, respectively.
The sequence of the orbitally decomposed spectral weights can be found in 'System.Name.out'.
In the spin-polarized calculation, 'System.Name.unfold_orbdn' is also generaged for the down spin case,
while only a single file 'System.Name.unfold_orb' is generated in the non-collinear calculation.
, energy relative to the chemical potential
in the unit of eV, and the orbitally decomposed spectral weights, respectively.
The sequence of the orbitally decomposed spectral weights can be found in 'System.Name.out'.
In the spin-polarized calculation, 'System.Name.unfold_orbdn' is also generaged for the down spin case,
while only a single file 'System.Name.unfold_orb' is generated in the non-collinear calculation.
As an example of plotting above the data by gnuplot, 'System.Name.unfold_plotexample' is generated. On the command line of the gnuplot, one can perform as
gnuplot> load `sic_primitive.unfold_plotexample'
gnuplot> set style data lines
gnuplot> set zeroaxis
gnuplot> set key below
gnuplot> set ytics 1
gnuplot> set mytics 5
gnuplot> set xra [0.000000:1.708883]
gnuplot> set yra [-10.0:6.0]
gnuplot> set ylabel "eV"
gnuplot> set xtics ("K" 0.000000, "G" 0.722258, "M" 1.347753, "K" 1.708882)
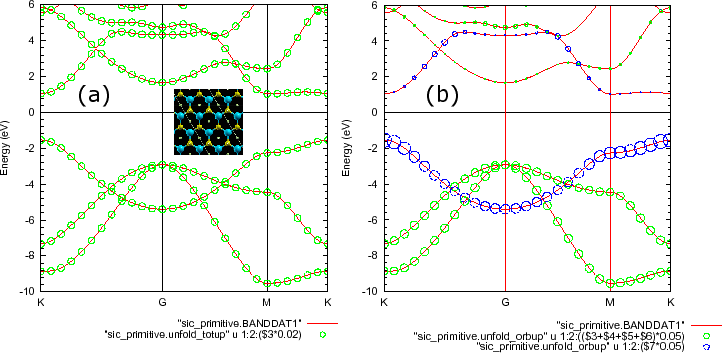
gnuplot> p "sic_primitive.BANDDAT1","sic_primitive.unfold_totup" u 1:2:($3*0.02) w circle
Now let's move on 'sic_primitive.unfold_orbup' storing orbitally decomposed spectral weights. In a similar way above, one can plot the orbitally decomposed spectral weights as
gnuplot> p "sic_primitive.BANDDAT1","sic_primitive.unfold_orbup" u 1:2:(($3+$4+$5+$6)*0.05) w circle, "sic_primitive.unfold_orbup" u 1:2:($7*0.05) w circle
 |