The specification of each keyword is given below. The list does not
include all the keywords in OpenMX, and those keywords will be explained
in each corresponding section.
File name
System.CurrrentDir
The output directory of output files is specified by this keyword.
The default is './'.
System.Name
The file name of output files is specified by this keyword.
DATA.PATH
The path to the VPS and PAO directories can be specified in your input file
by the following keyword:
DATA.PATH ../DFT_DATA13 # default=../DFT_DATA13
Both the absolute and relative specifications are available.
The default is '../DFT_DATA13'.
level.of.stdout
The amount of the standard output during the calculation is controlled by the keyword
'level.of.stdout'.
In case of 'level.of.stdout=0', minimum information.
In case of 'level.of.stdout=1', standard information.
In case of 'level.of.stdout=2', additional information together with
the minimum output information. 'level.of.stdout=3' is for developers.
The default is 1.
level.of.fileout
The amount of information output to the files is controlled by the keyword 'level.of.fileout'.
In case of 'level.of.fileout=0', minimum information (no Gaussian cube and grid files).
In case of 'level.of.fileout=1', standard output.
In case of 'level.of.fileout=2', additional information together with
the standard output. The default is 1.
Definition of Atomic Species
Species.Number
The number of atomic species in the system is specified by the
keyword 'Species.Number'.
Definition.of.Atomic.Species
Please specify atomic species by giving both the file name of pseudo-atomic
basis orbitals and pseudopotentials which must be existing
in the directories 'DFT_DATA13/PAO' and 'DFT_DATA13/VPS', respectively.
For example, they are specified as follows:
<Definition.of.Atomic.Species
H H5.0-s1>1p1>1 H_CA13
C C5.0-s1>1p1>1 C_CA13
Definition.of.Atomic.Species>
The beginning of the description must be '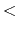 Definition.of.Atomic.Species', and
the last of the description must be 'Definition.of.Atomic.Species
Definition.of.Atomic.Species', and
the last of the description must be 'Definition.of.Atomic.Species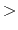 '.
In the first column, you can give any name to specify the atomic species.
The name is used in the specification of atomic coordinates by
'.
In the first column, you can give any name to specify the atomic species.
The name is used in the specification of atomic coordinates by
'Atoms.SpeciesAndCoordinates'.
In the second column, the file name of the pseudo-atomic basis orbitals
without the file extension and the number of primitive orbitals and
contracted orbitals are given. Here we introduce an abbreviation of
the basis orbital we used as H4.0-s11p11, where H4.0 indicates the file name
of the pseudo-atomic basis orbitals without the file extension which must
exist in the directory 'DFT_DATA13/PAO', s11 means that one optimized orbitals
are constructed from one primitive orbitals for the s-orbital, which means
no contraction. Also, in case of s11, corresponding to no contraction,
you can use a simple notation 's1' instead of 's11'. Thus, 'H4.0-s1p1' is
equivalent to 'H4.0-s11p11'.
In the third column, the file name for the pseudopotentials without
the file extension is given. Also the file must
exist in the directory 'DFT_DATA13/VPS'.
It can be possible to assign as the different atomic species for the
same atomic element by specifying the different basis orbitals and
pseudopotentials. For example, you can define the atomic species
as follows:
<Definition.of.Atomic.Species
H1 H5.0-s1p1 H_CA13
H2 H5.0-s2p2d1 H_CA13
C1 C5.0-s2p2 C_CA13
C2 C5.0-s2p2d2 C_CA13
Definition.of.Atomic.Species>
The flexible definition may be useful for the decrease of
computational efforts, in which only high level basis functions are
used for atoms belonging to the essential part which determines
the electric properties in the system, and lower level basis functions
are used for atoms in the other inert parts.
Atoms
Atoms.Number
The total number of atoms in the system is specified by the
keyword 'Atoms.Number'.
Atoms.SpeciesAndCoordinates.Unit
The unit of the atomic coordinates is specified by the keyword
'Atoms.SpeciesAndCoordinates.Unit'. Please specify 'Ang' when you
use the unit of Angstrom, and 'AU' when the unit of atomic unit.
The fractional coordinate is also available by 'FRAC'.
Then, please specify the coordinates spanned by
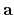 ,
, 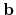 , and
, and 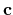 -axes given in 'Atoms.UnitVectors'.
In the fractional coordinates, the coordinates can range from -0.5 to 0.5,
and the coordinates beyond its range will be automatically adjusted
after the input file is read.
-axes given in 'Atoms.UnitVectors'.
In the fractional coordinates, the coordinates can range from -0.5 to 0.5,
and the coordinates beyond its range will be automatically adjusted
after the input file is read.
Atoms.SpeciesAndCoordinates
The atomic coordinates and the number of spin charge
are given by the keyword
'Atoms.SpeciesAndCoordinates' as follows:
<Atoms.SpeciesAndCoordinates
1 C 0.000000 0.000000 0.000000 2.0 2.0
2 H -0.889981 -0.629312 0.000000 0.5 0.5
3 H 0.000000 0.629312 -0.889981 0.5 0.5
4 H 0.000000 0.629312 0.889981 0.5 0.5
5 H 0.889981 -0.629312 0.000000 0.5 0.5
Atoms.SpeciesAndCoordinates>
The beginning of the description must be 'Atoms.SpeciesAndCoordinates', and
the last of the description must be 'Atoms.SpeciesAndCoordinates'.
The first column is a sequential serial number for identifying atoms.
The second column is given to specify the atomic species which
must be given in the first column of the specification of the keyword
'Definition.of.Atomic.Species' in advance. In the third, fourth, and fifth
columns, x-, y-, and z-coordinates are given.
When 'FRAC' is chosen for the keyword 'Atoms.SpeciesAndCoordinates.Unit',
the third, fourth, and fifth columns are fractional coordinates
spanned by , , and -axes, where the coordinates
can range from -0.5 to 0.5, and the coordinates beyond its range will be
automatically adjusted after the input file is read.
The sixth and seventh columns give
the number of initial charges for up and down spin states of each atom, respectively.
The sum of up and down charges must be the number of valence electrons for
the atomic element.
When you calculate spin-polarized systems using 'LSDA-CA' or 'LSDA-PW',
you can give the initial spin charges for each atom, which might be
those of the ground state, to accelerate the SCF convergence.
Atoms.UnitVectors.Unit
The unit of the vectors for the unit cell is specified by the keyword
'Atoms.UnitVectors.Unit'. Please specify 'Ang' when you
use the unit of Angstrom, and 'AU' when the unit of atomic unit.
Atoms.UnitVectors
The vectors, , , and of the unit cell are
given by the keyword 'Atoms.UnitVectors' as follows:
<Atoms.UnitVectors
10.0 0.0 0.0
0.0 10.0 0.0
0.0 0.0 10.0
Atoms.UnitVectors>
The beginning of the description must be 'Atoms.UnitVectors', and
the last of the description must be 'Atoms.UnitVectors'.
The first, second, and third rows correspond to the vectors, ,
, and of the unit cell, respectively.
If the keyword is absent in the cluster calculation, a unit cell is
automatically determined so that the isolated system cannot overlap
with the image systems in the repeated cells. See also the Section
'Automatic determination of the cell size'.
SCF or Electronic System
scf.XcType
The keyword 'scf.XcType' specifies the exchange-correlation potential.
Currently, 'LDA', 'LSDA-CA', 'LSDA-PW', and 'GGA-PBE' are available,
where 'LSDA-CA' is the local spin density functional of
Ceperley-Alder [2], 'LSDA-PW' is the local spin density
functional of Perdew-Wang, in which the gradient of density
is set to zero in their GGA formalism [4].
Note: 'LSDA-CA' is faster than 'LSDA-PW'.
'GGA-PBE' is a GGA functional proposed by Perdew et al [5].
scf.SpinPolarization
The keyword 'scf.SpinPolarization' specifies the non-spin polarization
or the spin polarization for the electronic structure.
If the calculation for the spin polarization is performed, then
specify 'ON'. If the calculation for the non-spin polarization
is performed, then specify 'OFF'. When you use 'LDA' for the keyword
'scf.XcType', the keyword 'scf.SpinPolarization' must be 'OFF'.
In addition to these options, 'NC' is supported for the non-collinear
DFT calculation. For this calculation, see also the Section
'Non-collinear DFT'.
scf.partialCoreCorrection
The keyword 'scf.partialCoreCorrection' is a flag for
a partial core correction (PCC) in calculations of exchange-correlation
energy and potential. 'ON' means that PCC is made, and 'OFF' is none.
In any cases, the flag should be 'ON', since pseudopotentials generated
with PCC should be used with PCC, and also PCC does not affect
the result for pseudopotentials without PCC because of zero PCC charge
in this case.
scf.Hubbard.U
In case of the LDA+U or GGA+U calculation, the keyword 'scf.Hubbard.U' should
be switched 'ON' (ON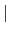 OFF). The default is 'OFF'.
OFF). The default is 'OFF'.
scf.Hubbard.Occupation
In the LDA+U method, three occupation number operators 'onsite',
'full', and 'dual' are available which can be specified by the
keyword 'scf.Hubbard.Occupation'.
Hubbard.U.values
An effective U-value on each orbital of species is defined by
the following keyword:
<Hubbard.U.values # eV
Ni 1s 0.0 2s 0.0 1p 0.0 2p 0.0 1d 4.0 2d 0.0
O 1s 0.0 2s 0.0 1p 0.0 2p 0.0 1d 0.0
Hubbard.U.values>
The beginning of the description must be 'Hubbard.U.values', and
the last of the description must be 'Hubbard.U.values'.
For all the basis orbitals specified by the 'Definition.of.Atomic.Species',
you have to give an effective U-value
in the above format. The '1s' and '2s' mean the first and second s-orbital,
and the number behind '1s' is the effective U-value (eV) for the first s-orbital.
The same rule is applied to p- and d-orbitals.
scf.Constraint.NC.Spin
The keyword 'scf.Constraint.NC.Spin' should be switched 'ON' (ONOFF)
when the constraint DFT method for the non-collinear spin orientation
is performed.
scf.Constraint.NC.Spin.v
The keyword 'scf.Constraint.NC.Spin.v' gives a prefactor (eV) of
the penalty functional in the constraint DFT for the non-collinear
spin orientation.
scf.ElectronicTemperature
The electronic temperature (K) is given by the keyword
'scf.ElectronicTemperature'. The default is 300 (K).
scf.energycutoff
The keyword 'scf.energycutoff' specifies the cutoff energy which is
used in the calculation of matrix elements associated with difference
charge Coulomb potential and exchange-correlation potential and
the solution of Poisson's equation using fast Fourier transform (FFT).
The default is 150 (Ryd).
scf.Ngrid
The keyword 'scf.Ngrid' gives the number of grids to discretize the
a-, b-, and c-axes.
Although 'scf.energycutoff' is usually used for the discretization,
the numbers of grids are specified by 'scf.Ngrid', they are used for
the discretization instead of those by 'scf.energycutoff'.
scf.maxIter
The maximum number of SCF iterations is specified by the keyword
'scf.maxIter'. The SCF loop is terminated at the number specified
by 'scf.maxIter' even if a convergence criterion is not satisfied.
The default is 40.
scf.EigenvalueSolver
The solution method for the eigenvalue problem is specified by
the keyword 'scf.EigenvalueSolver'. An O(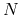 ) divide-conquer method 'DC',
an O() Krylov subspace method 'Krylov', a numerically exact low-order
scaling method 'ON2', the cluster calculation 'Cluster', and the band
calculation 'Band' are available.
) divide-conquer method 'DC',
an O() Krylov subspace method 'Krylov', a numerically exact low-order
scaling method 'ON2', the cluster calculation 'Cluster', and the band
calculation 'Band' are available.
scf.Kgrid
When you specify the band calculation 'Band' for the keyword
'scf.EigenvalueSolver', then you need to give a set of numbers (n1,n2,n3)
of grids to discretize the first Brillouin zone in the k-space by the keyword
'scf.Kgrid'. For the reciprocal vectors
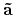 ,
,
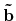 ,
and
,
and
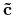 in the k-space, please provide a set of numbers
(n1,n2,n3) of grids as n1 n2 n3. The k-points in OpenMX are generated by a regular mesh method.
in the k-space, please provide a set of numbers
(n1,n2,n3) of grids as n1 n2 n3. The k-points in OpenMX are generated by a regular mesh method.
scf.ProExpn.VNA
Switch on the keyword 'scf.ProExpn.VNA' in case that the neutral atom potential VNA is expanded by
projector operators [31]. Otherwise turn off. The default is 'ON'.
scf.ProExpn.VNA ON # ON|OFF, default = ON
In case that 'scf.ProExpn.VNA=OFF', the matrix elements for the VNA potential are evaluated by
using the regular mesh in real space.
scf.Mixing.Type
A mixing method of the electron density (or the density matrix) to generate
an input electron density at the next SCF step is specified by keyword 'scf.Mixing.Type'.
A simple mixing method ('Simple'),
'GR-Pulay' method (Guaranteed-Reduction Pulay method) [41],
'RMM-DIIS' method [42],
'Kerker' method [43],
'RMM-DIISK' method [42]
and
'RMM-DIISH' method [42] are available.
The simple mixing method used here is modified to accelerate the convergence,
referring to a convergence history.
When 'GR-Pulay', 'RMM-DIIS', 'Kerker', 'RMM-DIISK', or 'RMM-DIISH' is used,
the following recipes are helpful to obtain faster convergence
of SCF calculations:
- Use a rather larger value for 'scf.Mixing.StartPulay'.
Before starting the Pulay-like mixing, achieve a convergence at some level.
An appropriate value may be 10 to 30 for 'scf.Mixing.StartPulay'.
- Use a rather larger value for 'scf.ElectronicTemperature'
in case of metallic systems. When 'scf.ElectronicTemperature' is small,
numerical instabilities appear often.
- Use a large value for 'scf.Mixing.History'.
In most cases, 'scf.Mixing.History=20' can be a good value.
Among these mixing schemes, the robustest one might be 'RMM-DIISK'.
scf.Init.Mixing.Weight
The keyword 'scf.Init.Mixing.Weight' gives the initial mixing weight used by
the simple mixing, the GR-Pulay, the RMM-DIIS, the Kerker, the RMM-DIISK, and the RMM-DIISH methods.
The valid range is 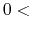 scf.Init.Mixing.Weight
scf.Init.Mixing.Weight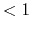 .
The default is 0.3.
.
The default is 0.3.
scf.Min.Mixing.Weight
The keyword 'scf.Min.Mixing.Weight' gives the lower limit of a mixing
weight in the simple and Kerker mixing methods.
The default is 0.001.
scf.Max.Mixing.Weight
The keyword 'scf.Max.Mixing.Weight' gives the upper limit of a mixing
weight in the simple and Kerker mixing methods.
The default is 0.4.
scf.Kerker.factor
The keyword gives a Kerker factor which is used in
the Kerker and RMM-DIISK mixing methods.
If the keyword is not given, a proper value is automatically determined.
For further details, see the Section 'SCF convergence'.
scf.Mixing.History
In the GR-Pulay method [41], the RMM-DIIS method [42],
the Kerker method [43], the RMM-DIISK method [42], and the RMM-DIISH method [42],
the input electron density (Hamiltonian) at the next SCF step is estimated based on
the output electron densities (Hamiltonian)
in the several previous SCF steps. The keyword 'scf.Mixing.History' specifies
the number of previous SCF steps which are used in the estimation.
For example,
if 'scf.Mixing.History' is specified to be 3, and when the SCF step is 6th,
the electron densities at 5, 4, and 3 SCF steps are
taken into account. Around 30 is a better choice.
scf.Mixing.StartPulay
The SCF step which starts the GR-Pulay, the RMM-DIIS,
the Kerker, the RMM-DIISK, and the RMM-DIISH methods is specified by the keyword
'scf.Mixing.StartPulay'. The SCF steps before
starting these Pulay-type methods are then performed by
the simple or Kerker mixing methods. The default is 6.
scf.Mixing.EveryPulay
The residual vectors in the Pulay-type mixing methods tend to become
linearly dependent each other as the mixing steps accumulate, and
the linear dependence among the residual vectors makes the convergence
difficult. A way of avoiding the linear dependence is to do the Pulay-type
mixing occasionally during the Kerker mixing.
With this prescription, you can specify the frequency using the
keyword 'scf.Mixing.EveryPulay'.
For example, in case of 'scf.Mixing.EveryPulay=5', the Pulay-mixing is
made at every five SCF iterations, while the Kerker mixing is used
at the other steps. 'scf.Mixing.EveryPulay=1' corresponds to the
conventional Pulay-type mixing. It is noted that the keyword
'scf.Mixing.EveryPulay' is supported for only 'RMM-DIISK', and
the default value is 1.
scf.criterion
The keyword 'scf.criterion' specifies a convergence criterion (Hartree)
for the SCF calculation. The SCF iteration is ended when a condition,
dUelescf.criterion, is satisfied, where dUele is defined as the
absolute deviation between the eigenvalue energy at the current and
previous SCF steps. The default is 1.0e-6 (Hartree).
scf.Electric.Field
give
The keyword 'scf.Electric.Field' gives the strength of a uniform external electric
field given by a sawtooth waveform. For example, when an electric field
of 1.0 GV/m (10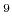 V/m) is applied along the a-axis, specify in your
input file as follows:
V/m) is applied along the a-axis, specify in your
input file as follows:
scf.Electric.Field 1.0 0.0 0.0 # default=0.0 0.0 0.0 (GV/m)
The sign of electric field is taken as that applied to electrons.
The default is 0.0 0.0 0.0.
scf.system.charge
The keyword 'scf.system.charge' gives the amount of the electron and
hole dopings. The plus and minus signs correspond to hole and electron
dopings, respectively. The default is 0.
scf.SpinOrbit.Coupling
When the spin-orbit coupling is included, the keyword should be 'ON',
otherwise please set to 'OFF'. In case of the inclusion of the spin-orbit
coupling, you have to use j-dependent pseudopotentials.
See also the Section 'Relativistic effects' as for the j-dependent
pseudopotentials.
1D FFT
1DFFT.EnergyCutoff
The keyword '1DFFT.EnergyCutoff' gives the energy range to tabulate
the Fourier transformed radial functions of pseudo-atomic orbitals and
of the projectors for non-local potentials.
The default is 3600 (Ryd).
1DFFT.NumGridK
The keyword '1DFFT.NumGridK' gives the the number of radial grids
in the k-space. The values of the Fourier transformation for radial functions
of pseudo-atomic orbitals and of the projectors for non-local potentials
are tabulated on the grids, ranging from zero to 1DFFT.EnergyCutoff,
as a function of radial axis in the k-space.
The default is 900.
1DFFT.NumGridR
The keyword '1DFFT.NumGridR' gives the the number of radial grids
in real space which is used in the numerical grid integrations of the
Fourier transformation for radial functions of pseudo-atomic orbitals and of the
projectors for non-local potentials.
The default is 900.
Orbital Optimization
orbitalOpt.Method
The keyword 'orbitalOpt.Method' specifies a method for the orbital
optimization. When the orbital optimization is not performed, then
choose 'OFF'. When the orbital optimization is performed, the following
two options are available: 'atoms' in which basis orbitals on each atom are
fully optimized, 'species' in which basis orbitals on each species are optimized.
In 'atoms', the radial functions of basis orbitals are optimized
with a constraint that the radial wave function 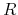 is independent
on the magnetic quantum number, which guarantees the rotational invariance
of the total energy. However, the optimized orbital on all the atoms can
be different from each other.
In the 'species', basis orbitals in atoms with the same species name,
that you define in 'Definition.of.Atomic.Species', are optimized as the
same orbitals. If you want to assign the same orbitals to atoms with almost
the same chemical environment, and optimize these orbitals, this scheme
is useful.
is independent
on the magnetic quantum number, which guarantees the rotational invariance
of the total energy. However, the optimized orbital on all the atoms can
be different from each other.
In the 'species', basis orbitals in atoms with the same species name,
that you define in 'Definition.of.Atomic.Species', are optimized as the
same orbitals. If you want to assign the same orbitals to atoms with almost
the same chemical environment, and optimize these orbitals, this scheme
is useful.
orbitalOpt.scf.maxIter
The maximum number of SCF iterations in the orbital optimization is
specified by the keyword 'orbitalOpt.scf.maxIter'.
orbitalOpt.Opt.maxIter
The maximum number of iterations for the orbital optimization is specified
by the keyword 'orbitalOpt.Opt.maxIter'. The iteration loop for the orbital
optimization is terminated at the number specified by 'orbitalOpt.Opt.maxIter'
even if a convergence criterion is not satisfied.
orbitalOpt.Opt.Method
Two schemes for the optimization of orbitals are available:
'EF' which is an eigenvector following method, 'DIIS' which is
the direct inversion method in iterative subspace.
The algorithms are basically the same as for the geometry optimization.
Either 'EF' or 'DIIS' is chosen by the keyword 'orbitalOpt.Opt.Method'.
orbitalOpt.StartPulay
The quasi Newton method 'EF' and 'DIIS' starts from the optimization step
specified by the keyword 'orbitalOpt.StartPulay'.
orbitalOpt.HistoryPulay
The keyword 'orbitalOpt.HistoryPulay' specifies the number of previous steps
to estimate the next input contraction coefficients used in the quasi Newton
method 'EF' and 'DIIS'.
orbitalOpt.SD.step
The orbital optimization at optimization steps before moving to the quasi Newton method 'EF' or 'DIIS'
is performed by the steepest decent method. The prefactor used in the steepest decent method is specified
by the keyword 'orbitalOpt.SD.step'. In most cases, 'orbitalOpt.SD.step' of 0.001 can be a good prefactor.
orbitalOpt.criterion
The keyword 'orbitalOpt.criterion' specifies a convergence criterion
((Hartree/Borg)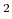 ) for the orbital optimization. The iterations loop is
finished when a condition, Norm of derivativesorbitalOpt.criterion,
is satisfied.
) for the orbital optimization. The iterations loop is
finished when a condition, Norm of derivativesorbitalOpt.criterion,
is satisfied.
CntOrb.fileout
If you want to output the optimized radial orbitals to files,
then the keyword 'CntOrb.fileout' must be 'ON'.
Num.CntOrb.Atoms
The keyword 'Num.CntOrb.Atoms' gives the number of atoms whose
optimized radial orbitals are output to files.
Atoms.Cont.Orbitals
The keyword 'Atoms.Cont.Orbitals' specifies the atom number,
which is given by the first column in the specification of
the keyword 'Atoms.SpeciesAndCoordinates' for the output
of optimized orbitals as follows:
<Atoms.Cont.Orbitals
1
2
Atoms.Cont.Orbitals>
The beginning of the description must be 'Atoms.Cont.Orbitals', and
the last of the description must be 'Atoms.Cont.Orbitals'.
The number of lines should be consistent with the number specified
in the keyword 'Atoms.Cont.Orbitals'. For example, the name of files
are C_1.pao and H_2.pao, where the symbol corresponds to that
given by the first column in the specification of the keyword
'Definition.of.Atomic.Species' and the number after the symbol means
that of the first column in the specification of the keyword
'Atoms.SpeciesAndCoordinates'. These output files 'C_1.pao' and 'H_2.pao'
can be an input data for pseudo-atomic orbitals as is.
SCF Order-N
orderN.HoppingRanges
The keyword 'orderN.HoppingRanges' defines the radius of a sphere which is
centered on each atom. The physically truncated cluster for each atom is
constructed by picking up atoms inside the sphere with the radius in the DC and Krylov
subspace O() methods.
orderN.KrylovH.order
The dimension of the Krylov subspace of Hamiltonian in each truncated cluster
is given by the 'orderN.KrylovH.order'.
orderN.KrylovS.order
In case of 'orderN.Exact.Inverse.S=off', the inverse is approximated
by a Krylov subspace method for the inverse, where the dimension of
the Krylov subspace of overlap matrix in each truncated cluster is
given by the keyword 'orderN.KrylovS.order'.
The default value is 'orderN.KrylovH.order'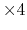 .
.
orderN.Exact.Inverse.S
In case of 'orderN.Exact.Inverse.S=on', the inverse of overlap
matrix for each truncated cluster is exactly evaluated.
Otherwise, see the keyword 'orderN.KrylovS.order'.
The default is 'on' (onoff).
orderN.Recalc.Buffer
In case of 'orderN.Recalc.Buffer=on', the buffer matrix is recalculated
at every SCF step. Otherwise, the buffer matrix is calculated at
the first SCF step, and fixed at subsequent SCF steps.
The default is 'on' (onoff).
orderN.Expand.Core
In case of 'orderN.Expand.Core=on', the core region
is defined by atoms within a sphere with radius of
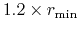 , where
, where
 is the distance between the central atom and the nearest atom.
The core region defines a set of vectors used for the first step in the generation
of the Krylov subspace for each truncated cluster.
In case of 'orderN.Expand.Core=off', the central atom is considered
as the core region. The default is 'on' (onoff).
is the distance between the central atom and the nearest atom.
The core region defines a set of vectors used for the first step in the generation
of the Krylov subspace for each truncated cluster.
In case of 'orderN.Expand.Core=off', the central atom is considered
as the core region. The default is 'on' (onoff).
MD or Geometry Optimization
MD.Type
Please specify the type of the molecular dynamics calculation or the geometry optimization.
Currently, NO MD (Nomd), MD with the NVE ensemble (NVE), MD with the NVT ensemble by
a velocity scaling scheme (NVT_VS)[19],
MD with the NVT ensemble by a Nose-Hoover scheme (NVT_NH) [20],
MD with multi-heat bath (NVT_VS2 or NVT_VS4),
the geometry optimization by the steepest decent (SD) method (Opt),
DIIS optimization method (DIIS),
the eigenvector following (EF) method (EF) [47],
and the rational function (RF) method (RF) [48]
are available.
For the details, see the Sections 'Geometry optimization' and 'Molecular dynamics'.
MD.Fixed.XYZ
In the geometry optimization and the molecular dynamics simulations,
it is possible to separately fix the x-, y-, and z-coordinates of the
atomic position to the initial position in your input file by
the following keyword:
<MD.Fixed.XYZ
1 1 1 1
2 1 0 0
MD.Fixed.XYZ>
The example is for a system consisting of two atoms. If you have N atoms,
then you have to provide N rows in this specification. The 1st column is
the same sequential number to specify atom as in the specification of the keyword
'Atoms.SpeciesAndCoordinates'. The 2nd, 3rd, and 4th columns are flags for the x-, y-, and
z-coordinates, respectively. '1' means that the coordinate is fixed, and '0' relaxed.
In the above example, the x-, y-, and z-coordinates of the atom '1' are fixed,
only the x-coordinate of the atom '2' is fixed. The default setting is that all the
coordinates are relaxed. The fixing of atomic positions are valid all the geometry
optimizers and molecular dynamics schemes.
MD.maxIter
The keyword 'MD.maxIter' gives the number of MD iterations.
MD.TimeStep
The keyword 'MD.TimeStep' gives the time step (fs).
MD.Opt.criterion
When any of the geometry optimizers is chosen for the keyword 'MD.Type',
then the keyword 'MD.Opt.criterion' specifies a convergence criterion
(Hartree/Bohr). The geometry optimization is finished when a condition,
the maximum force on atom is smaller than 'MD.Opt.criterion', is satisfied.
MD.Opt.DIIS.History
The keyword 'MD.Opt.DIIS.History' gives the number of previous
steps to estimate the optimized structure used in the geometry
optimization by 'DIIS', 'EF', and 'RF'. The default value is 3.
MD.Opt.StartDIIS
The geometry optimization step at which 'DIIS', 'EF', or 'RF' starts is specified
by the keyword 'MD.Opt.StartDIIS'.
The geometry optimization steps before starting the DIIS-type method
is performed by the steepest decent method.
The default value is 5.
MD.TempControl
The keyword specifies temperature for atomic motion in
MD of the NVT ensembles.
In 'NVT_VS', the temperature for nuclear motion can be controlled by
<MD.TempControl
3
100 2 1000.0 0.0
400 10 700.0 0.4
700 40 500.0 0.7
MD.TempControl>
The beginning of the description must be 'MD.TempControl', and
the last of the description must be 'MD.TempControl'.
The first number '3' gives the number of the following lines
to control the temperature. In this case, you can see that there
are three lines. Following the number '3', in the consecutive
lines the first column means MD steps and the second column gives
the interval of MD steps that the velocity scaling is made.
For the above example, a velocity scaling is performed at every
two MD steps until 100 MD steps, at every 10 MD steps from 100
to 400 MD steps, and at every 40 MD steps from 400 to 700 MD steps.
The third and fourth columns give a given temperature (K) and
a scaling parameter 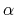 in the interval. For further details
see the Section 'Molecular dynamics'.
On the other hand, in NVT_NH, the temperature for nuclear motion
can be controlled by
in the interval. For further details
see the Section 'Molecular dynamics'.
On the other hand, in NVT_NH, the temperature for nuclear motion
can be controlled by
<MD.TempControl
4
1 1000.0
100 1000.0
400 700.0
700 600.0
MD.TempControl>
The beginning of the description must be 'MD.TempControl', and
the last of the description must be 'MD.TempControl'.
The first number '4' gives the number of the following lines to
control the temperature. In this case you can see that there are
four lines. Following the number '4', in the consecutive lines
the first and second columns give the MD steps and a given temperature
for nuclear motion. The temperature between the MD steps explicitly specified by
the keyword is given by linear interpolation.
NH.Mass.HeatBath
In 'NVT_NH', a mass of heat bath is given by the keyword.
The default mass is 20, where we use the unified atomic mass unit
that the principal isotope of carbon atom is 12.0.
MD.Init.Velocity
For molecular dynamics simulations, it is possible to provide the initial
velocity of each atom by the following keyword:
<MD.Init.Velocity
1 3000.000 0.0 0.0
2 -3000.000 0.0 0.0
MD.Init.Velocity>
The example is for a system consisting of two atoms. If you have N atoms,
then you have to provide N rows in this specification.
The 1st column is the same sequential number to specify atom as in the
specification of the keyword 'Atoms.SpeciesAndCoordinates'.
The 2nd, 3rd, and 4th columns are x-, y-, and z-components of the velocity of
each atom. The unit of the velocity is m/s.
The keyword 'MD.Init.Velocity' is compatible with the keyword 'MD.Fixed.XYZ'.
Band dispersion
Band.dispersion
When you evaluate the band dispersion, please specify 'ON' for the keyword
'Band.dispersion'.
Band.KPath.UnitCell
The keyword 'Band.KPath.UnitCell' gives unit vectors, which are
used in the calculation of the band dispersion, as follows:
<Band.KPath.UnitCell
3.56 0.0 0.0
0.0 3.56 0.0
0.0 0.0 3.56
Band.KPath.UnitCell>
The beginning of the description must be 'Band.KPath.UnitCell', and
the last of the description must be 'Band.KPath.UnitCell'.
If 'Band.KPath.UnitCell' exists, the reciprocal lattice vectors for
the calculation of the band dispersion are calculated by
the unit vectors specified in 'Band.KPath.UnitCell'.
If 'Band.KPath.UnitCell' is not found, the reciprocal lattice
vectors, which are calculated by the unit vectors specified in
'Atoms.UnitVectors', is employed for the calculation of the
band dispersion. In case of fcc, bcc, base centered cubic,
and trigonal cells, the reciprocal lattice vectors
for the calculation of the band dispersion should be specified
using the keyword 'Band.KPath.UnitCell' based on the consuetude
in the band calculations.
Band.Nkpath
The keyword 'Band.Nkpath' gives the number of paths for the band
dispersion.
Band.kpath
The keyword 'Band.kpath' specifies the paths of the band dispersion
as follows:
<Band.kpath
15 0.0 0.0 0.0 1.0 0.0 0.0 g X
15 1.0 0.0 0.0 1.0 0.5 0.0 X W
15 1.0 0.5 0.0 0.5 0.5 0.5 W L
15 0.5 0.5 0.5 0.0 0.0 0.0 L g
15 0.0 0.0 0.0 1.0 1.0 0.0 g X
Band.kpath>
The beginning of the description must be 'Band.kpath', and
the last of the description must be 'Band.kpath'.
The number of lines should be consistent with 'Band.Nkpath'.
The first column is the number of grids at which eigenvalues
are evaluated on the path. The following (n1, n2, n3) and
(n1', n2', n3'), spanned by the reciprocal lattice vectors,
specifies the starting and ending k-points of the path in the first
Brillouin zone.
If 'Band.KPath.UnitCell' is found, the reciprocal lattice vectors for
the calculation of the band dispersion are calculated by
the unit vectors specified in 'Band.KPath.UnitCell'.
If 'Band.KPath.UnitCell' is not found, the reciprocal lattice
vectors, which are calculated by the unit vectors specified in
'Atoms.UnitVectors' is employed for the calculation of the
band dispersion.
The final two alphabets give the name of the starting and
ending k-points of the path.
Restarting
scf.restart
If you want to restart the SCF calculation using a previous file 'System.Name_rst/*'
which should be generated in the previous calculation, then set the keyword 'scf.restart' to
'ON'.
Output of molecular orbitals (MOs)
MO.fileout
If you want to output molecular orbitals (MOs) to files, then set
the keyword 'MO.fileout' to 'ON'.
num.HOMOs
The keyword 'num.HOMOs' gives the number of
the highest occupied molecular orbitals (HOMOs)
that you want to output to files.
num.LUMOs
The keyword 'num.LUMOs' gives the number of
the lowest unoccupied molecular orbitals (LUMOs)
that you want to output to files.
MO.Nkpoint
When you have specified 'MO.fileout=ON' and
'scf.EigenvalueSolver=Band', the keyword 'MO.Nkpoint' gives
the number of the k-points at which you output MOs
to files.
MO.kpoint
The keyword 'MO.kpoint' specifies the k-point, at which MOs are evaluated
for the output to files, as follows:
<MO.kpoint
0.0 0.0 0.0
MO.kpoint>
The beginning of the description must be 'MO.kpoint', and
the last of the description must be 'MO.kpoint'.
The k-points are specified by (n1, n2, n3) which is spanned
by the reciprocal lattice vectors, where the the reciprocal
lattice vectors are determined in the same way as 'Band.kpath'.
DOS and PDOS
Dos.fileout
If you want to evaluate density of states (DOS) and projected
partial density of states (PDOS), please set in 'Dos.fileout=ON'.
Dos.Erange
The keyword 'Dos.Erange' determines the energy range for
the DOS calculation as
Dos.Erange -10.0 10.0
The first and second values are the lower and upper bounds of
the energy range (eV) for the DOS calculation, respectively.
Dos.Kgrid
The keyword, Dos.Kgrid, gives a set of numbers (n1,n2,n3)
of grids to discretize the first Brillouin zone in the k-space,
which is used in the DOS calculation.
Interface for developers
HS.fileout
If you want to use Kohn-Sham Hamiltonian, overlap, and density matrices,
please set in 'HS.fileout=ON'. Then, these data are stored to 'System.Name.scfout' in a
binary form, where 'System.Name' is the file name specified by the keyword 'System.Name'.
The utilization of these data is illustrated in the Section 'Interface for developers'.
Voronoi charge
Voronoi.charge
If you want to calculate Voronoi charges, then set
the keyword 'Voronoi.charge' in 'ON'. The result is found in 'System.Name.out',
'System.Name' is the file name specified by the keyword 'System.Name'.
2016-04-03