Next: Cluster calculation Up: MPI parallelization Previous: MPI parallelization Contents Index
When the O(![]() ) method is employed, it is expected that one can obtain
a good parallel efficiency because of the inherent algorithm.
A typical MPI execution is as follows:
) method is employed, it is expected that one can obtain
a good parallel efficiency because of the inherent algorithm.
A typical MPI execution is as follows:
% mpirun -np 4 openmx DIA512_DC.dat > dia512_dc.std &
The input file 'DIA512_DC.dat' found in the directory 'work' is
for the SCF calculation (1 MD) of the diamond including 512 carbon
atoms using the divide-conquer (DC) method.
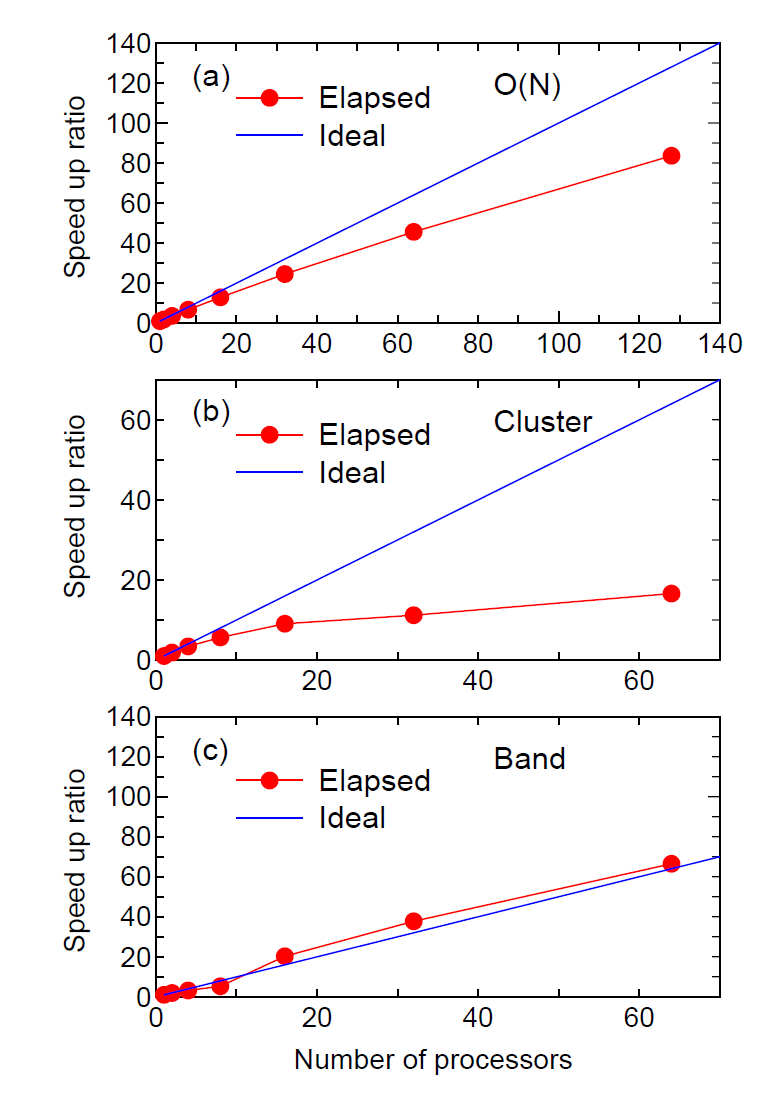
The speed-up ratio in comparison of the elapsed time per MD step
is shown in Fig. 21 (a) as a function of the number of processes
on a CRAY-XC30 (2.6 GHz/Xeon processors).
We see that the parallel efficiency decreases as the number of
processors increase, and the speed-up ratio at 128 CPUs is
about 84. The decreasing efficiency is due to the decrease of
the number of atoms allocated to one processor.
So, the weight of other unparallelized parts such as disk I/O becomes
significant. Moreover, it should be noted that the efficiency is
significantly reduced in non-uniform systems in terms of atomic
species and geometrical structure due to disruption of the road
balance, while an algorithm is implemented to avoid the disruption.
See also the subsection 'Krylov subspace method' for further information on
parallelization.
 |
2016-04-03