Efficient implementation of the nonequilibrium Green function method for electronic transport calculations
The original has been published
in Phys. Rev. B 81, 035116 (2010).
Abstract
An efficient implementation of the nonequilibrium Green function (NEGF) method combined with the density functional theory (DFT) using localized pseudo-atomic orbitals (PAOs) is presented for electronic transport calculations of a system connected with two leads under a finite bias voltage. In the implementation, accurate and efficient methods are developed especially for evaluation of the density matrix and treatment of boundaries between the scattering region and the leads. Equilibrium and nonequilibrium contributions in the density matrix are evaluated with very high precision by a contour integration with a continued fraction representation of the Fermi-Dirac function and by a simple quadrature on the real axis with a small imaginary part, respectively. The Hartree potential is computed efficiently by a combination of the two dimensional fast Fourier transform (FFT) and a finite difference method, and the charge density near the boundaries is constructed with a careful treatment to avoid the spurious scattering at the boundaries. The efficiency of the implementation is demonstrated by rapid convergence properties of the density matrix. In addition, as an illustration, our method is applied for zigzag graphene nanoribbons, a Fe/MgO/Fe tunneling junction, and a LaMnOSrMnO superlattice, demonstrating its applicability to a wide variety of systems.
1 INTRODUCTION
The non-equilibrium Green function (NEGF) method[1, 2, 3, 4, 5, 6] potentially has several advantages to investigate electronic transport properties of nano-scale materials such as single molecules,[7, 8] atomic wires,[9, 10] carbon based materials,[11, 12] and thin layers.[13, 14] The potential advantages are summarized by the following features of the NEGF method: (i) the source and drain contacts are treated based on the same theoretical framework as for the scattering region. [3, 4, 6] (ii) the electronic structure of the scattering region under a finite source-drain bias voltage is self-consistently determined by combining with first principle electronic structure calculation methods such as the density functional theory (DFT) and the Hartree-Fock (HF) method. [17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27] (iii) many body effects in the transport properties, e.g., electron-phonon[28, 29, 30, 31, 32, 33] and electron-electron interactions,[34, 35, 36, 37] might be included through self-energies without largely deviating the theoretical framework. (iv) its applicability to large-scale systems can be anticipated, since the NEGF method relies practically on the locality of basis functions in real space, resulting in computations for sparse matrices.[15] Due to those potential advantages, recently several groups have implemented the NEGF method coupled with the DFT or HF method using atomic-type or the other local basis functions with successful applications for calculations of the electronic transport properties. [15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27]
However, a highly accurate and efficient implementation method must be still developed from the following two reasons: The first obvious reason is to extend the applicability of the NEGF method to large-scale systems. The efficient implementation might lead to more challenging applications of the NEGF method to very large-scale complicated systems. The majority part in the computational effort of the NEGF method mainly comes from the evaluation of the density matrix which is decomposed into the evaluation of Green functions and numerical integrations. Thus, the efficient calculation of the part is a key factor for extending the applicability to large-scale systems. Nevertheless, accurate and efficient methods for evaluating density matrix within the NEGF method have not been fully developed, although several methods have been already proposed. [17, 18, 38] To extend the applicability of the NEGF method to large-scale systems, a remarkably efficient method that we have recently developed[45] will be applied for the problem in this study, and we will show that the new method is much faster than the other method.[18] The second reason is that spurious scattering should be negligible when the NEGF method is extended to include the many body effects beyond the one particle picture.[28, 29, 30, 31, 32, 33, 34, 35, 36, 37] The spurious scattering accompanied by the inaccurate implementation might make the many body effects indistinct in the electronic transport properties. One can imagine that the spurious scattering can be easily produced in the NEGF method, since unlike the conventional band structure calculations NEGF has to be evaluated by a patch work that the self consistent field (SCF) calculations of the source and drain leads are performed beforehand, and the calculated results are incorporated in the NEGF calculations through the self energy and the boundary conditions between the scattering region and leads. Therefore, a careful treatment to handle the boundary conditions should be developed to avoid the spurious scattering.
In this paper, to address the above two issues we present an accurate and efficient implementation of the NEGF method, in combination with DFT using pseudo-atomic orbitals (PAOs) and pseudopotentials, using a contour integration method which is based on a continued fraction representation of the Fermi-Dirac function. For the accurate treatment of the boundary conditions between the scattering region and leads, we also develop a method for calculating the Hartree potential by a combination of the two dimensional fast Fourier transform (FFT) and a finite difference method so that the boundary condition can be correctly reproduced. In addition, we discuss a careful treatment to construct the charge density near the boundaries. The efficiency and accuracy of our implementation are demonstrated by several numerical test calculations on convergence of the density matrix.
This paper is organized as follows: In Sec. 2 the details of our implementation for treating the equilibrium state of the scattering region are discussed by focusing on the evaluation of the equilibrium density matrix, the treatment for constructing the charge density near the boundaries, and an efficient method for calculating the Hartree potential. In Sec. 3 our implementation for the nonequilibrium state is described. In Sec. 4 we demonstrate the accuracy and efficiency of the implementation by a series of numerical calculations and several applications. In Sec. 5 our implementation of the NEGF method is summarized.
2 EQUILIBRIUM STATE
Since most of practical aspects in the implementation of the NEGF method coupled with DFT using the localized PAOs[42, 43, 44] appear in the ground state calculation of the system at equilibrium, we start our discussion from the electronic structure calculation of the equilibrium ground state by using the Green function method with DFT.
2.1 Equilibrium Green function (EGF)
Let us consider a system where one dimensional infinite cells are arranged with two dimensional periodicity as shown in Fig. 1. Throughout the paper, we assume that the electronic transport along the a-axis is of interest, and that the two dimensional periodicity spreads over the bc-plane. The one dimensional infinite cell consists of the central region denoted by and the cells denoted by and , where . All the cells and , arranged semi-infinitely, contain the same number of atoms with the same structural configuration, respectively, but the cells and can be different from each other. In the equilibrium state with a common chemical potential everywhere in the system, the electronic structure of the system may be determined by DFT.[40, 41, 65]
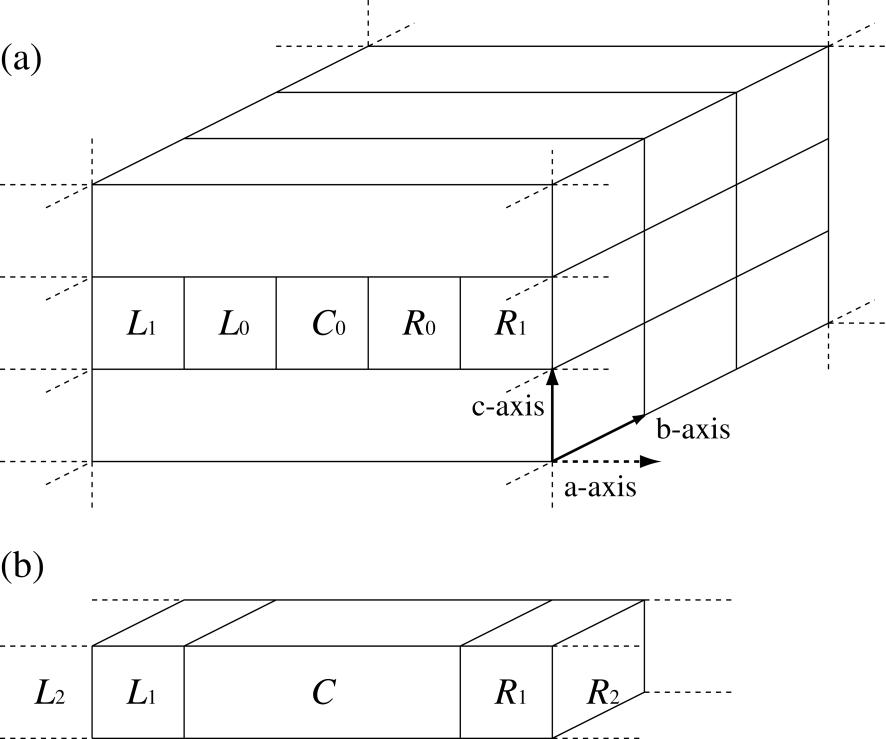
Figure 1: (a) Configuration of the system, treated by the NEGF method, with infinite left and right leads along the a-axis under a two dimensional periodic boundary condition on the bc-plane. (b) One dimensional system compacted from the configuration of (a) by considering the periodicity on the bc-plane, where the region is an extended central region consisting of , , and .
Due to the periodicity of the bc-plane the one-particle Kohn-Sham (KS) wave function in the system is expressed by the Bloch function on the bc-plane using PAOs located on site as:
| (1) |
where , , , and are indices for the spin, eigenstate, site, and basis orbital, respectively. The lattice vector and the Bloch wave vector are given by , where and are the lattice vectors, and , where and are the reciprocal lattice vectors, respectively. The summation over and is considered for all the basis orbitals in the one dimensional infinite cell, which indicates no periodicity along the -axis. Considering the variation of the total energy, within the conventional DFT, of the system expressed by the KS wave function Eq. (1) with respect to coefficients we obtain the following KS matrix equation:
| (2) |
where is a column vector consisting of the coefficients . The Hamiltonian and overlap matrices are given by
| (3) | |||||
| (4) |
where and are the Hamiltonian and overlap matrix elements between two basis functions and , respectively. The overlap matrix arises from the non-orthogonality of the PAO basis functions.[42, 43, 44] Now we consider an extended central region composed of the regions , , and as shown in Figs. 1 (a) and (b). The extension of the central region is made so that the relaxation of electronic structure around the interfaces between the leads, and , and the central region can be allowed. In addition, we impose two conditions:
-
(i)
The localized basis orbitals in the region overlap with those in the regions and , but do not overlap with those in the regions and .
-
(ii)
The localized basis orbitals in the () region has no overlap with basis orbitals in the cells beyond the nearest neighboring cells () and ().
In our implementation the basis functions are strictly localized in real space because of the generation of basis orbitals by a confinement scheme.[42, 43, 44] Therefore, once the localized basis orbitals with specific cutoff radii are chosen for each region, the two conditions can be always satisfied by just adjusting the size of the unit cells for and . This is a benefit in the use of the strictly localized basis orbitals compared to other local basis orbitals such as Slater- and Gaussian-type orbitals. In the use of the strictly localized basis orbitals, the eigenvalue problem in the Hilbert space spanned by the basis orbitals are solved without introducing any cutoff scheme. On the other hand, in the Green function method the matrix elements of the Hamiltonian and overlap matrices have to be truncated so as to satisfy the above two conditions in case of the other localized basis orbitals with the small but long tail. With the above two conditions (i) and (ii), the Hamiltonian matrix given by Eq. (3) is written by a block tridiagonal form as follows:
| (5) |
where , , and are Hamiltonian matrices of the central , left and right regions of which matrix size are the same as the number of basis orbitals , , and in the regions , , and , respectively. The other block components in Eq. (5) are the Hamiltonian matrices connecting two regions among the regions, and these matrix sizes are deduced from those of the two regions. Also the completely same structure is found in the overlap matrix. Thus, the electronic structure of the system given by Fig. 1(a) can be obtained by solving the one dimensional block chain model, being -dependent, given by Fig. 1(b) and the corresponding Eq. (2). By noting and making use of the block tridiagonal form of the Hamiltonian and overlap matrices, the Green function of the central region can be written by
| (6) |
with self energies and defined by
| (7) | |||||
| (8) |
where and are surface Green functions of the left and right regions.
It is worth pointing out that there is an energy functional which can be variationally minimized with respect to charge density if Eq. (6) is self-consistently solved. The details of derivation for the functional is given in Appendix A.
2.2 Surface Green function
In general, the surface Green function is defined by , where and are the Hamiltonian and overlap matrices for the lead regions, and the suffix, , is or . It is noted that due to the two conditions (i) and (ii) mentioned above the Hamiltonian and overlap matrices for the lead regions can be written by a block tridiagonal form as follows:
| (9) | |||||
| (10) |
where the Hamiltonian can be spin and -dependent, while the indices for them are omitted for simplification of the notation, and also the index appearing in corresponds to the cell number for the lead or . It seems to be difficult to directly diagonalize the KS equation Eq. (2) for the one dimensional block chain model because of the infinite dimension of the matrices. However, instead by focusing on only the central region one can evaluate the Green function of the central region as a rather small problem of in size. The effect of semi infinite regions and are included through the corresponding self energies and . In order to practically calculate the Green function of the central region given by Eq. (6), we introduce an approximation where the regions are all equivalent to each other with respect to the spatial charge distribution, the KS Hamiltonian, and the relevant density matrix which are calculated in advance by adopting the system of which unit cell is and by using the conventional band structure calculation. The same approximation also applies for the regions . Strictly speaking, the assumption is not correct, since the charge distribution must be affected by the interaction between the central region and the regions (). However, if the size of the unit vector along the -axis for the regions and in the extended central region is large enough, the assumption will be asymptotically correct as the unit vector becomes larger. The approximation enables us to evaluate the surface Green function by the iterative method.[46] The efficient iterative scheme can be performed by the following procedure:
| (11) | |||||
| (12) | |||||
| (13) | |||||
| (14) | |||||
| (15) | |||||
| (16) |
with a set of initial values
| (17) | |||||
| (18) | |||||
| (19) | |||||
| (20) |
By the iterative calculation, in most cases the inverse of rapidly converges at a part of the surface Green function:
| (21) |
where is the (1,1) block element of the surface Green function . The (1,1) block element of (or ) in size has all the necessary information to calculate the self-energies and , since there is no contribution from the other block elements because of the two conditions (i) and (ii) mentioned above. In practice, the convergence in the iterative calculation is very fast and the Frobenius norm, defined by , of (eV) is obtained by typically only 7 iterations.
2.3 Equilibrium density matrix
One of practical difficulties in the implementation of the Green function method is how the equilibrium density matrix is evaluated efficiently and accurately.[17, 18, 38, 47, 48, 49, 50, 51] In our implementation, the equilibrium density matrix is highly efficiently computed using the contour integration method with a special treatment of the Fermi-Dirac function .[45] If the Hamiltonian and overlap matrices associated with Eq. (6) are -dependent, it turns out that the spectrum function in the Lehmann representation of the central Green function is complex number in general as discussed in Appendix B of Ref. [45]. Then, the density matrix , where one of the associated basis orbitals is in the central cell and the other is in the cell denoted by , is given by making use of both the retarded and advanced Green functions and as
| (22) |
with
| (23) |
where is the volume of the unit cell, represents the integration over the first Brillouin zone, a positive infinitesimal, and a chemical potential. The integration over -space is numerically performed by using the Monkhorst-Pack mesh.[57] It is also noted that the phase factor appears through Eq. (1). If the Hamiltonian and overlap matrices are -independent, Eq. (22) can be simplified into a well-known formula:
| (24) |
In case of the -independent problem, the simplified formula is used, since the number of the evaluation of the Green function is reduced by half.
For the efficient integration in Eq. (23), in our implementation the Fermi-Dirac function is expressed by a continued fraction representation derived from a hypergeometric function[45] so that the structure of poles can be suitable for the integration associated with the Green function as follows:
| (25) | |||||
where with , is electronic temperature, and are complex variables. Also, and are the poles and the associated residues of the continued fraction representation Eq. (25), which are obtained via an eigenvalue problem derived from Eq. (25).[45] Since all the are real numbers, the poles are located on the imaginary axis. One may find an interesting distribution of the poles on the complex plane that the interval between neighboring poles are uniformly located up to about 61 % of the total number of poles on the half complex plane with the same interval , and from then onward it increases very rapidly as the distance between the pole and the real axis increases. The structure of the poles in Eq. (25) allows us to efficiently evaluate Eq. (23) because of the asymptotic change, , of the Green function in the faraway region of the real axis. In other words, the denser poles are allocated for the rapidly varying range of the Green function, and the coarser for smoothly. In addition, there is no ambiguity in the choice of the path in the contour integration unlike the other schemes,[17, 18, 38, 66] which is one of advantages in our method. By terminating the summation of Eq. (25) at a finite number of poles , the integration of Eq. (23) can be performed by the contour integration where the poles in the upper and lower half planes are taken into account for the terms with plus and minus signs in Eq. (23), respectively, and the explicit formula is given by
| (26) |
where , and is the zeroth order moment of the Green function . The zeroth order moment is easily calculated by which can be derived from the moment representation of the Green function,[45] where is a large real number and in this study (eV) is used in order to make the higher order moments negligible. Although the number of poles in the summation of Eq. (26) required for the sufficient convergence depends on the electronic temperature , the fully convergent result within double precision is achieved by the use of only 100 poles in case of as shown later.
If forces on atoms are calculated based on the conventional DFT scheme using the non-orthogonal basis orbitals,[18] the evaluation of the energy density matrix is needed. In Appendix B, we derive the calculation scheme of the equilibrium energy density matrix based on the contour integration method.
2.4 Charge density near the boundary
Even though the basis functions we used are strictly localized in real space, there is the nonnegligible contribution for the charge density near the boundary between the central and lead regions from the basis functions located in the lead regions. Note that any treatment for the contribution to the charge density has not been clarified in the other implementations.[17, 18, 19, 20, 21, 22, 23] Thus, we carefully calculate the charge density in the central region by considering three contributions:
| (27) |
where the suffix, s, is or , and , , and are the charge densities contributed from the basis functions located in the central, the lead and central, and the lead regions, respectively. Note that the summation over s is not required in Eq. (27) because of the conditions (i) and (ii). Each charge contribution is explicitly given by
| (28) | |||||
| (29) | |||||
| (30) |
where a is the lattice vector of the unit cell for the or region along the a-axis. The displacement of () denotes that the basis function is placed in the () region in the configuration shown in Fig. 1. The charge density given by Eq. (28) is calculated by the equilibrium density matrix given by Eq. (22). Although Eq. (28) can give a finite electron density on the outside of the central cell with because of the overlap of basis functions, the contribution is reflected in the central cell with by considering the periodicity on the bc-plane. In the nonequilibrium case, the equilibrium density matrix is only replaced by the nonequilibrium density matrix which will be discussed in the next section. Each term in the summations for the two contributions and survive only if the overlap of the associated two basis orbitals is not zero in the central region. Since the original central region is extended by adding the and regions, it is expected that the density matrix elements and in Eqs. (29) and (30) are close to those of the leads in the equilibrium condition. Therefore, the density matrix elements of the leads calculated by the conventional band structure calculations are used for Eqs. (29) and (30). Due to the treatment the charge densities and are independent of the SCF iteration so that for the computational efficiency they can be computed on a numerical mesh and stored before the SCF iteration. The factor 2 for in Eq. (27) is due to taking account of the contribution from , while the factor does not appear for , since all the paired terms are included by the double summation in Eq. (30).
We add a note that the same consideration has to be applied even for the calculation of the density of states (DOS). In this case, the contribution from off-diagonal block Green functions connecting the central and lead regions should be added to the DOS of the central region calculated by . The off-diagonal block Green functions can be calculated from and the surface Green functions, and , by making use of the identity as follows:
| (31) |
where s is or .
2.5 Hartree potential with the boundary condition
The Hartree potential in the central region is calculated under the boundary condition that the Hartree potential at the boundary between the central and regions is same as that of the lead, where the Hartree potential in both the lead regions is calculated using the conventional band structure calculation before the calculation of the infinite in Fig. 1(b) using the Green function. In our implementation, the Hartree potential for the central region with the boundary condition is efficiently evaluated by a combination of the two dimensional FFT and a finite difference method, while other schemes are used in the other implementations.[17, 18] The majority part of the Hartree potential in our treatment is accurately calculated by considering the neutral atom potential which is the sum of the local potential of the pseudopotential and the Hartree potential by the confined charge for the neutralization.[52] The neutral atom potential depends on only the atomic structure and atomic species, and has no relation with the boundary condition. The effect of the relaxation of charge distribution on the Hartree potential is taken into account by the remaining minority part of the Hartree potential given by
| (32) |
where is defined by the difference between the electron density calculated by the Green function and the atomic electron density[53] calculated by superposition of each atomic electron density at atomic site as follows:
| (33) |
The Fourier transformation of Eq. (32) on the yz plane, corresponding to the bc-plane depicted in Fig. 1(a), yields
| (34) |
where with integer numbers and . By approximating the second derivative in Eq. (34) with the simplest finite difference,
we obtain a simultaneous linear equation of in size with a tridiagonal matrix defined by
| (36) |
and a vector defined by
| (37) |
where is the interval between neighboring points and , runs from to , and and are the boundary conditions. Since the lattice vector of the extended central region along the a-axis is divided by for the discretization, the positions and are situated at the boundary, along the a-axis, of the unit cell of the central region . Thus, and can be calculated from the Hartree potential at the left boundaries along the a-axis of the left and right leads, respectively. After solving the simultaneous linear equations for all the points, the inverse two dimensional FFT on the bc-plane yields the difference Hartree potential under the boundary conditions. It is apparent that there is no ambiguity for the inclusion of the boundary conditions in the method. Although the treatment can be easily extended to the higher order finite difference to the second derivative, we restrict ourselves to the simplest case in the implementation because of its sufficient accuracy. Employing FFT for two dimensional Fourier transformation, the whole computational effort to solve the Poisson equation Eq. (32) under the boundary conditions is estimated to be which is slightly superior to that of the three dimensional FFT, where , , and are the number of meshes for the discretization along the -, -, and -axes.
2.6 Hamiltonian and overlap matrix elements
Our implementation of the NEGF method with DFT is based on the strictly localized PAOs[42, 43, 44] and a norm-conserving pseudopotential method.[59] Within the scheme, the calculation of the matrix elements such as the overlap and kinetic energy integrals, consisting of two center integrals, is performed using a Fourier transform method,[60] while the other matrix elements for and , which cannot be decomposed into two center integrals, are evaluated by the numerical integration on the regular mesh in real space.[63] The further details on how the elements of the Hamiltonian and overlap matrices are calculated can be found in Ref. [52].
In addition to the above evaluation of the Hamiltonian and overlap matrix elements, the Hamiltonian matrix elements associated with the basis orbitals situated at near the boundary are treated in a special way as explained below. If the tails of two basis orbitals located on atoms in the central region go beyond the boundary between the central and the lead regions, the associated Hamiltonian matrix element is replaced by the corresponding element in the lead region calculated by the conventional band structure calculation. The case can happen only if the two basis orbitals are located in the region () because of the condition (i) so that the replacement of the Hamiltonian matrix element can be always possible. The replacement is made by assuming that the potential profile near the boundary is similar to that near the boundary between the regions and , and can be justified if the size of the region is large enough.
2.7 Charge mixing
Compared to conventional band structure calculations, it seems that the NEGF method tends to suffer from difficulty in obtaining the SCF convergence. Our observation in several cases suggests that the difficulty may come from charge sloshing along the a-axis during the SCF iteration. The difference Hartree potential change largely by imposition of the boundary condition even for a small variation in the charge density distribution, resulting in a serious charge sloshing along the a-axis. Thus, we consider suppression of the charge sloshing along the a-axis by introducing the following weight factor :
| (38) |
where is a smaller one of either or , and and are adjustable parameters, while keeping . The prefactor is given by
| (39) |
with definitions:
| (40) | |||||
| (41) |
where is an adjustable parameter. Noting that is the number of difference electron density of each layer indexed by , and that the Coulomb potential induced by each charged layer depends linearly on the distance from the layer, one can notice that is proportional to the electric field at position . Therefore, Eq. (38) takes charge density under a large electric field into significant account in addition to the suppression of the charge sloshing in the bc-plane in a sense by the Kerker method.[54] In our implementation, the weight factor given by Eq. (38) is combined with the Kerker method[54] and the residual minimization method in a direct inversion iterative subspace (RMM-DIIS)[55] with substantial improvement.
A technical remark should also be added to avoid a local trap problem in the SCF calculation. In systems having a long a-axis, a unphysical charge distribution, corresponding to a large charge separation in real space, tends to be obtained even after achieving the self consistency. In this case the Hamiltonian of the central region at the first SCF iteration, which are calculated via superposition of atomic charge density, is far from the self-consistently converged one, while the Hamiltonian matrix used in the calculation for the self-energy is determined in a self-consistent manner beforehand. The inconsistency between the two matrices tends to produce a unphysical charge distribution at the first SCF iteration. Once the situation happens at the first SCF iteration, in many cases the electronic structure keeps trapped during the subsequent SCF iteration, which is a serious problem in practical applications. However, the local trap problem can be overcome by a simple scheme that the first few SCF iterations are performed by using the conventional band scheme and then onward the solver is switched from the band scheme to the NEGF method. In the band structure calculation for the first few iterations, such a unphysical charge distribution does not appear due to no self-energy involved. In most cases we find that the simple scheme works well to avoid the local trap problem in the SCF convergence.
3 NONEQUILIBRIUM STATE
3.1 Nonequilibrium density matrix
Based on the NEGF theory mainly developed by Schwinger[1] and Keldysh[2], the density matrix in the nonequilibrium state of the central region is evaluated by[17, 18, 21, 22]
| (42) |
In addition to the equilibrium density matrix given by Eq. (22), a correction term defined by
| (43) |
is taken into account, where is defined by
| (44) |
with
| (45) |
and
| (46) |
Either the left or right chemical potential which is lower than the other is used for the calculation of the equilibrium density matrix in Eq. (42). As well, in Eqs. (44) and (46) the chemical potentials are given by the rule that and if , and and if . Starting from the NEGF theory, the formula, Eq. (42), may be derived by introducing two assumptions. The first assumption is that the occupation of the wave functions incoming from the left (right) region still obeys the Fermi-Dirac function with the left (right) chemical potential even in the central region. The assumption can be justified within at least the one-particle picture, since the same result can be obtained from the Lippmann-Schwinger equation for a non-interacting system.[5, 17, 39] The second assumption is that in the central region the states in the energy regime below the lower chemical potential is in equilibrium due to the other physical obstacles such as the electron-phonon[28, 29, 30, 31, 32, 33] and electron-electron interactions,[34, 35, 36, 37] which are not considered explicitly in our implementation, although the definite role of those obstacles is obscure as for the occupation of the states. The second assumption allows electrons to occupy in highly localized states below the lower chemical potential through the first term of Eq. (42). Only the states in the energy regime between two chemical potentials are treated as in the nonequilibrium condition in which the contribution of the wave functions incoming from the lead with the higher chemical potential is taken into account to form the correction term given by Eq. (43).
The integrand in Eq. (44) is not analytic apart from the real axis, since the integrand is a function of both and . Thus, one cannot apply the contour integration method that we use for the equilibrium density matrix. Instead, a simple rectangular quadrature scheme is applied to the integration of Eq. (44) on the real axis with a small imaginary part . Since the integrand contains the difference between two Fermi-Dirac functions, the energy range for the integration can be effectively reduced to a narrow range that the difference is larger than a threshold, where the threshold of is used in this study. With the threshold and the step width of 0.01 (eV), the number of meshes on the real axis is 152 for (eV) at . The convergence speed depends on the shape of the integrand and how large is employed for smearing the integrand, which will be discussed later.
3.2 Source-drain and gate bias voltages
The source-drain bias voltage applied to the left and right leads is easily incorporated by adding a constant electric potential to the Hartree potential in the right lead region. The effect of the bias voltage appears at three places. The first effect is that the Hamiltonian matrix in the right region given by Eq. (9) is replaced using Eq. (10) as
| (47) |
This can be easily confirmed by noting that the matrix elements for the constant potential is . The off-diagonal block elements , , and appearing Eqs. (7) and (8) are also replaced in the same way. The second effect is that the chemical potential of the right lead is replaced as
| (48) |
The treatment is made so that the first replacement can be regarded as just shifting the origin of energy in the right lead. The last effect is that the boundary condition in Eq. (37) is replaced as
| (49) |
where is calculated by the Fourier transformation on the bc-plane for at plus . Since only the difference of the bias voltages applied to the left and right leads affects the result, one can consider the replacements on only the right lead at the three places as shown above. It is noted that the replacement by Eq. (49) corresponds to adding a linear potential to the Hartree potential in the central region , where and are determined by the boundary conditions: and .
In our implementation, the gate voltage is treated by adding an electric potential defined by
| (50) |
where is a constant value corresponding to the gate voltage, the center of the region , and the length of the unit vector along a-axis for the region . Due to the form of Eq. (50), the applied gate voltage affects mainly the region in the central region . The electric potential may resemble the potential produced by the image charges.[56]
3.3 Transmission and current
The spin resolved transmission is evaluated by the Landauer formula for the non-interacting central region connected with two leads:
| (51) |
where is the spin and -resolved transmission defined by
| (52) |
Using the transmission formula, the current is evaluated by
| (53) |
The formula can be derived by starting from a more general formula of the current for the interacting central region and by replacing the involved Green functions with the non-interacting Green functions as shown by Meir and Wingreen.[58] We perform the integration in Eq. (53) on the real axis with a small imaginary part by the same way as for the nonequilibrium density matrix of Eq. (44).
4 NUMERICAL RESULTS
4.1 Computational details
All the calculations in this study were performed by the DFT code, OpenMX.[64] The PAOs centered on atomic sites are used as basis functions.[42, 43, 44] The PAO basis functions we used, generated by a confinement scheme,[42, 43] are specified by H5.5-s2, C4.5-s2p2, O5.0-s2p2d1, Fe5.0-s2p2d1, Mg5.5-s2p2, La7.0-s3p2d1f1, Sr7.0-s3p2d1f1, and Mn6.0-s3p2d2, where the abbreviation of basis functions, such as C4.5-s2p2, represents that C stands for the atomic symbol, 4.5 the cutoff radius (bohr) in the generation by the confinement scheme, and s2p2 means the employment of two primitive orbital for each of s- and p-orbitals. Norm-conserving pseudopotentials are used in a separable form with multiple projectors to replace the deep core potential into a shallow potential.[59] Also, a local density approximation (LDA) to the exchange-correlation potential is employed.[61], while a generalized gradient approximation (GGA)[62] is used only for calculations of the LaMnOSrMnO superlattice. The real space grid techniques are used with the cutoff energies of 120-200 Ry in numerical integrations and the solution of Poisson equation using FFT.[63] In addition, the projector expansion method is employed in the calculation of three-center integrals for the deep neutral atom potentials.[52]
4.2 Convergence properties
The accuracy and efficiency of the implementation are mainly determined by the evaluation of density matrix given by Eq. (42) which consists of two contributions: the equilibrium and non-equilibrium terms given by Eqs. (22) and (43), respectively. In this subsection, we discuss the convergence properties of the equilibrium and non-equilibrium terms in the density matrix as a function of numerical parameters.
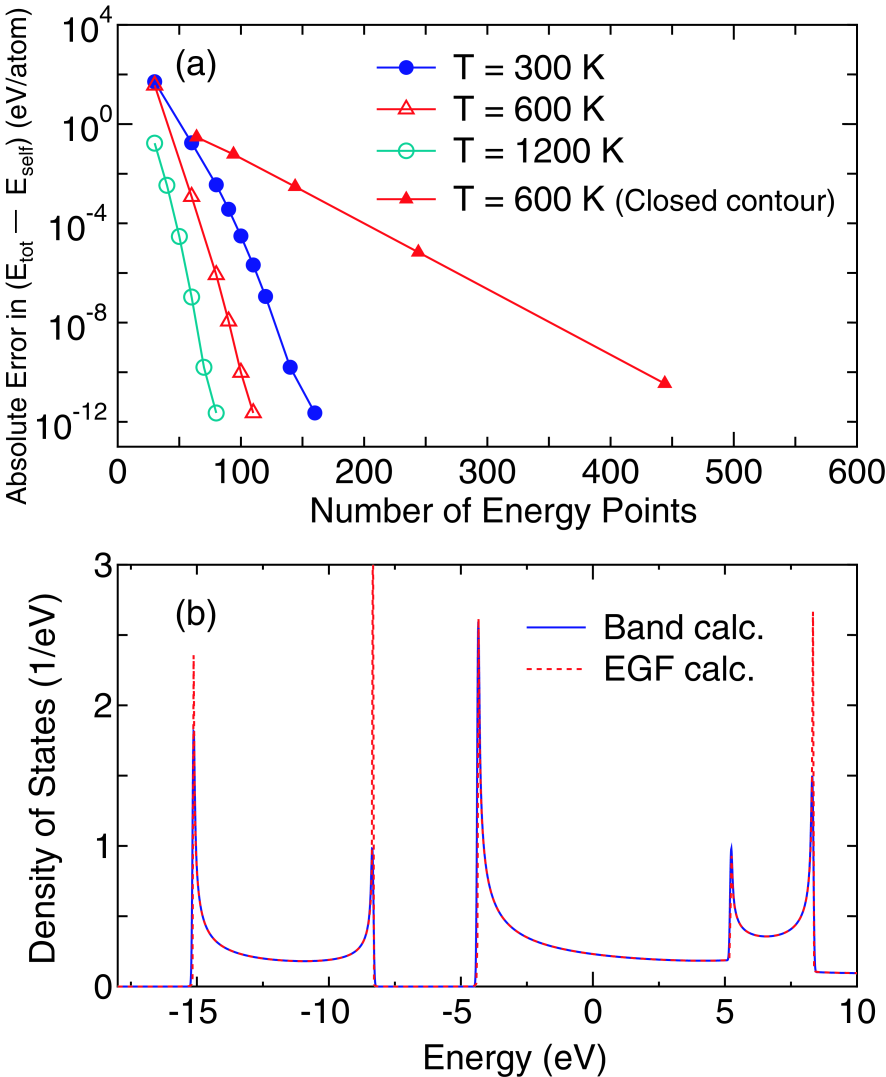
Figure 2: (Color online)(a) Absolute error in () per atom of a linear carbon chain with a bond length of 1.4 Å under the zero bias voltage at electronic temperature of 300, 600, and 1200 K, where the regions , , and contain four carbon atoms, respectively. For comparison the same calculation (closed contour) using a closed contour method[18, 67] is also shown for T=600 K. The definitions of and are found in Appendix A. The reference values are obtained from calculations with a large number of poles. (b) Total DOS of the carbon linear chain, calculated by the conventional band structure calculation (solid line) and the EGF method (dotted line), under the zero bias voltage at 300 K, where 160 poles are used for the integration of the equilibrium density matrix. It is hard to distinguish two lines due to the nearly equivalent results.
Since the majority part of the density matrix given by Eq. (42) is the equilibrium contribution, let us first discuss convergency of the equilibrium density matrix as a function of poles. The absolute error in of a carbon linear chain is shown in Fig. 2(a) as a function of the number of poles in order to illustrate the convergence property for the equilibrium density matrix under zero bias voltage, where can be regarded as a conventional expression of the total energy in DFT, and the definitions of the two energy terms and with the Fermi-Dirac function are given in Appendix A. For comparison the result calculated by a closed contour method is also shown.[18, 67] One can see that the accuracy of eV per atom is obtained using 140, 100, and 70 poles for the electronic temperature of 300, 600, and 1200 K, respectively, while about 400 energy points are needed to obtain the same accuracy using the closed contour method at 600 K.[18] For most cases, we find that the convergence rate is similar to the case shown in Fig. 2(a). In general, the number of poles to achieve the accuracy of eV per atom must be proportional to the inverse of , since the interval between neighboring poles of the continued fraction given by Eq. (25) is scaled by . This fact implies that the computational effort increases as the electronic temperature decreases. However, we generally use electronic temperature from 300 to 1000 K for practical calculations, which means that the use of 100 poles is enough for practical purposes. Therefore, it can be concluded that the most contribution of the density matrix can be very accurately evaluated with a small number of poles, i.e., 100.
To demonstrate the proper treatment of the boundary between the lead and the central regions in our implementation, in Fig. 2(b) we show a comparison between the conventional band structure and the EGF calculations with respect to DOS of the carbon linear chain. The comparison provides a severe test to check whether the EGF method is properly implemented or not. It can be confirmed that DOS calculated by the EGF method is nearly equivalent to that by the conventional band structure calculation, which clearly shows the proper treatment of the boundary between the lead and the central regions in our scheme.
As explained before, the integration of Eq. (44) required for the evaluation of the nonequilibrium term in the density matrix has to be performed on the real axis with a small imaginary part because contour integration schemes may not be applied due to the non-analytic nature of the integrand. The treatment might suffer from numerical instabilities in the SCF iteration, since the integrand can rapidly vary due to the existence of poles of Green function located on the real axis. A remedy to avoid the numerical problem is to smear the Green function by introducing a relatively large imaginary part.[17, 18, 21, 22]
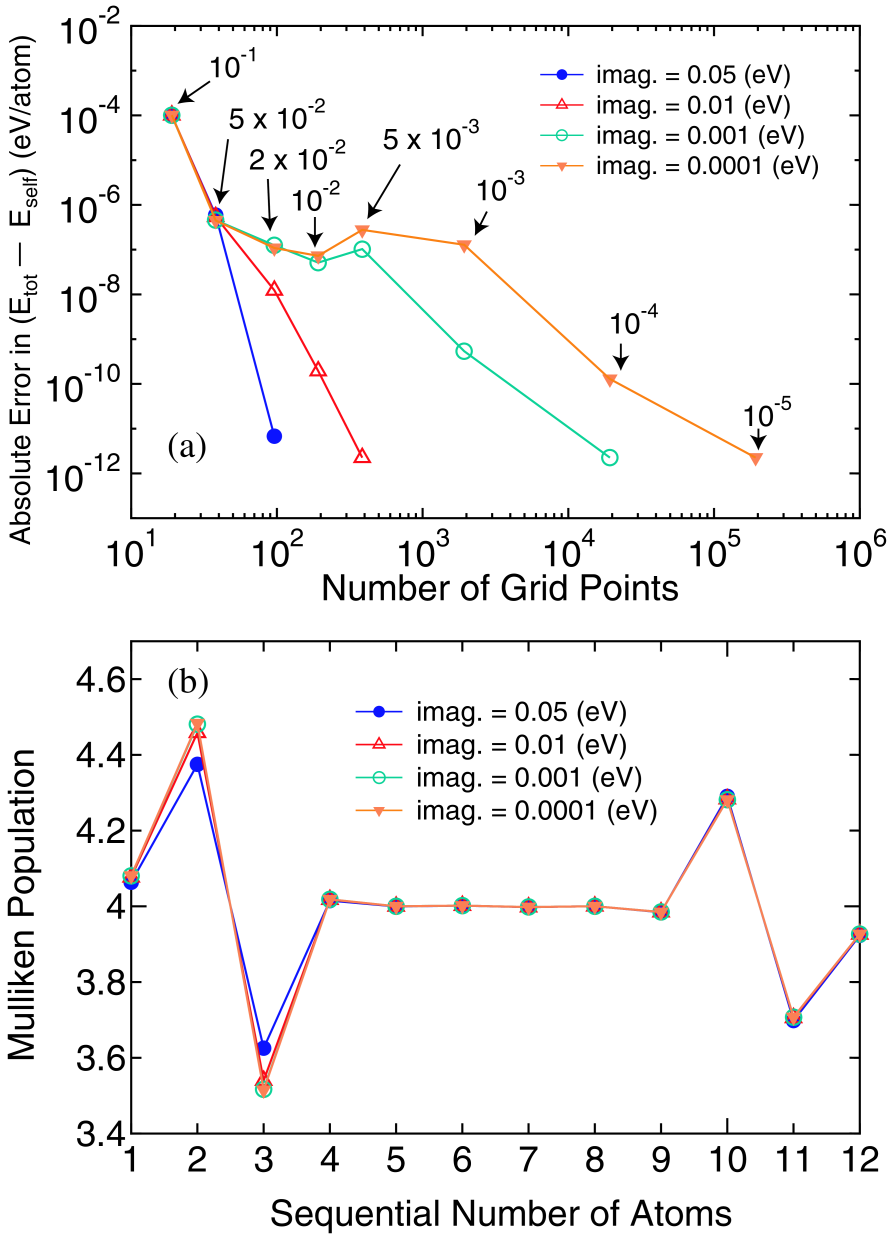
Figure 3: (Color online) (a) Absolute error in () per atom of the same linear carbon chain as in Fig. 2, calculated by various imaginary parts, under a finite bias voltage of 0.5 V, where the other calculation conditions are same as for Fig. 2(b). The number pointed by the arrow denotes a grid spacing (eV) corresponding to the number of grid points. The reference values are obtained from calculations with a large number of grid points. (b) Mulliken populations in the carbon chain. The sequential numbers 1 and 12 correspond to the most left and right hand side atoms in the central region , respectively.
To investigate convergency of the nonequilibrium term in the density matrix, in Fig. 3(a) we show the absolute error in of the same infinite carbon chain as in Fig. 2 but under a finite bias voltage of 0.5 eV as a function of the number of regular grid points used for the evaluation of the nonequilibrium term in the density matrix given by Eqs. (43) and (44). We also tested the Gauss-Legendre quadrature for the integration of the nonequilibrium term besides the integration using the regular grid, but found that the convergence rate of the Gauss-Legendre quadrature is rather slower than the simple scheme possibly due to the spiky structure of the integrand. Thus, we have decided to use the simple scheme using the regular grid. As expected, it turns out that the number of grid points to achieve the accuracy of eV per atom increases as the imaginary part becomes smaller. However, the accuracy of eV is attainable using about 100 grid points in case of the imaginary part of 0.01 eV, while a few thousands grid points have to be used to achieve the same accuracy for the imaginary part of 0.0001 eV.
Although the accuracy of eV can be achieved by introducing the smearing scheme, however, one may consider that results can be affected by the introduction of an imaginary part. In order to find a compromise between the accuracy and efficiency, the Mulliken population of the carbon linear chain under the finite bias voltage of 0.5 eV is shown in Fig. 3(b). We see that the use of the imaginary part of 0.01 eV gives a result comparable to that obtained by the use of 0.0001 eV. Thus, the imaginary part of 0.01 eV can be a compromise between the accuracy and efficiency in this case. As a result, one can find that the energy points of 200 (100 and 100 for the equilibrium and nonequilibrium density matrices, respectively) are enough to achieve the accuracy of eV per atom in at 600 K. However, it should be mentioned that a proper choice of the imaginary part must depend on the electronic structure of systems, and the careful consideration must be taken into account especially for a case that the spiky DOS appears in between two chemical potentials of the leads, while for the equilibrium part of the density matrix the convergence property is insensitive to the electronic structure.
We also note that the accurate evaluation of the density matrix makes the SCF calculation stable even under a finite bias voltage. In fact, for the case with the bias voltage of 0.5 V the number of the SCF iterations to achieve the residual norm of for the charge density difference is 29 which is nearly equivalent to that, 30, for the zero bias case.
4.3 Interpolation of the effect by the bias voltage
Since for large-scale systems it is very time-consuming to perform the SCF calculation at each bias voltage, here we propose an interpolation scheme to reduce the computational cost in the calculations by the NEGF method. The interpolation scheme is performed in the following way: (i) the SCF calculations are performed for a few bias voltages which are selected in the regime of the bias voltage of interest. (ii) when the transmission and current are calculated, a linear interpolation is made for the Hamiltonian block elements, and , of the central scattering region and the right lead, and the chemical potential, , of the right lead by
| (54) | |||||
| (55) | |||||
| (56) |
where the indices and in the superscript mean that the quantities are calculated or used at the corresponding bias voltages where the SCF calculations are performed beforehand. Note that it is also possible to perform the interpolation for -independent Hamiltonian matrix elements instead of Eqs. (54) and (55). In general, should range from 0 to 1 for the moderate interpolation. A comparison between the fully self consistent and the interpolated results is shown with respect to the current and transmission in the linear carbon chain in Figs. 4(a) and (b). In this case, the SCF calculations at three bias voltages of 0, 0.5, and 1.0 V are performed, and the results at the other bias voltages are obtained by the interpolation scheme. For comparison we also calculate the currents via the SCF calculations at all the bias voltages. It is confirmed that the simple interpolation scheme gives notably accurate results for both the calculations of the current and transmission. Although the proper selection of bias voltages used for the SCF calculations may depend on systems, the result suggests that the simple scheme is very useful to interpolate the effect of the bias voltage while keeping the accuracy of the calculations.
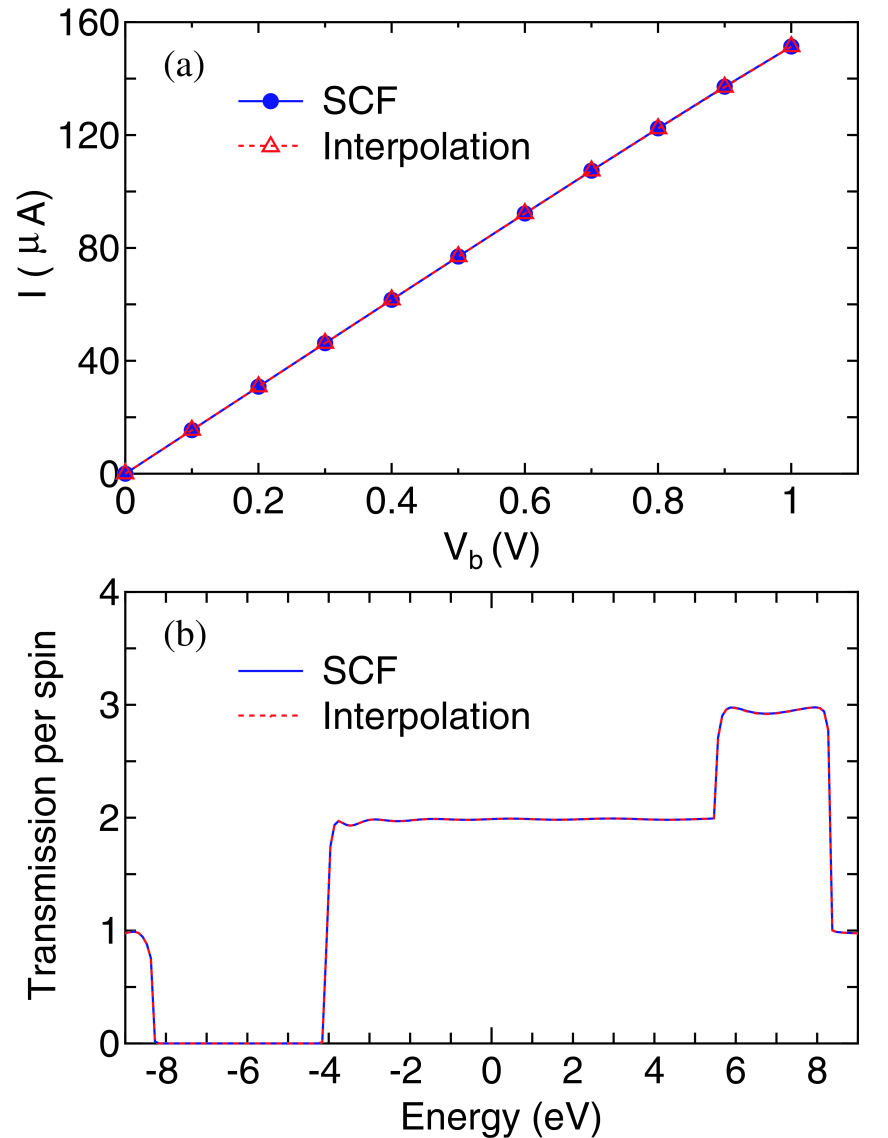
Figure 4: (Color online) (a) Currents of the linear carbon chain calculated by the SCF calculations (solid line) and the interpolation scheme (dotted line). (b) Transmission of the linear carbon chain under a bias voltage of 0.3 V, calculated by the SCF calculations (solid line) and the interpolation scheme (dotted line). The imaginary part of 0.01 and the grid spacing of 0.01 eV are used for the integration of the nonequilibrium term in the density matrix. The other calculation conditions are same as for Fig.2(b).
4.4 Applications
4.4.1 Zigzag graphene nanoribbons
As an illustration of our implementation, we investigate transport properties of zigzag graphene nanoribbons (ZGNRs) with different magnetic configurations. A characteristic feature in the band structure of ZGNR is the appearance of flat bands around X-point near the Fermi level, resulting in spin-polarization of associated states located at the zigzag edges.[68, 69] Thus, so far several intriguing transport properties have been theoretically predicted especially for ZGNRs among GNRs by focusing on the spin polarized edge states.[70, 11, 12, 71, 72, 73, 74, 75, 76, 77] For instance, it is found that ZGNRs might exhibit an extraordinary large magnetoresistance (MR) effect and a spin polarized current.[12]
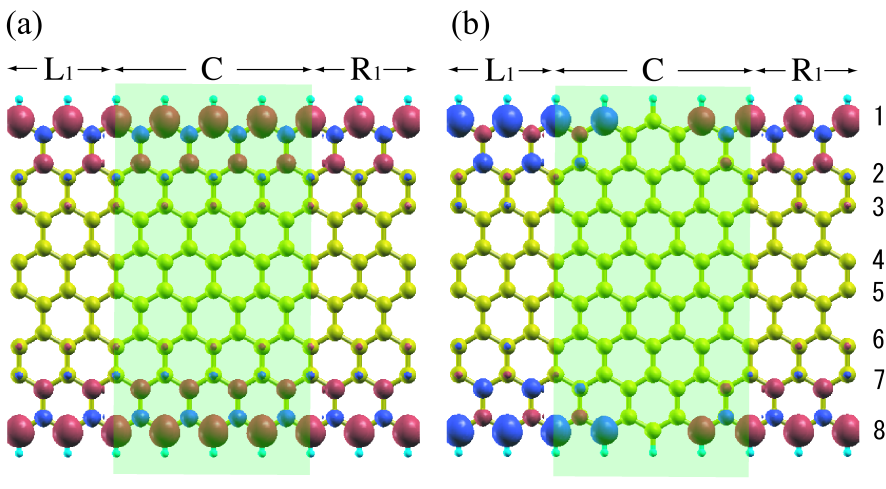
Figure 5: (Color online) 8-ZGNR with (a) a FM junction and (b) an AFM junction together with the spatial distribution of the spin density at the source-drain bias voltage V. The zigzag edges are terminated by hydrogen atoms. The isosurface value of is used for drawing the spin density.
Here we focus on the current-bias voltage (-) characteristic of - and -ZGNRs with two magnetic configurations: ferromagnetic (FM) and antiferromagnetic (AFM) junctions as shown in Figs. 5(a) and (b), respectively, where the number, or , is the number of carbon atom in the sublattice being across ZGNR along the lateral direction. The odd and even cases will also be referred to as asymmetric and symmetric, respectively. The extended central region consists of one sublattice and four unit cells, and contains 72 and 82 atoms for 7- and 8-ZGNRs, respectively. The poles of 100 is used for the evaluation of the equilibrium density matrix with the electronic temperature of 300 K, while the nonequilibrium term in the density matrix is evaluated using the simple quadrature method with the imaginary part of 0.01 eV and the grid spacing of 0.02 eV. The geometric structures used are optimized under the periodic boundary condition until the maximum force is less than hartree/bohr. At each bias voltage the electronic structure of ZGNR is self-consistently determined.
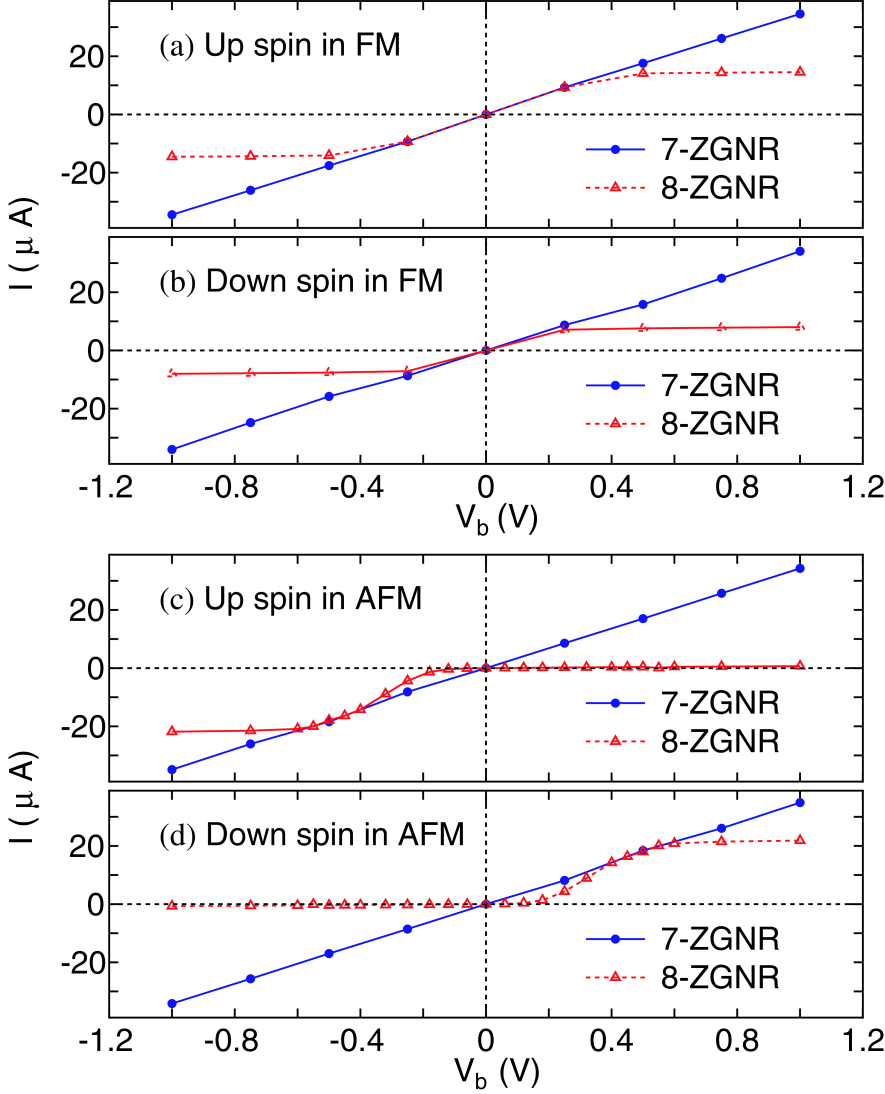
Figure 6: (Color online) - curves for (a) the up-spin and (b) the down-spin states in the FM junction, and (c) the up-spin and (d) the down-spin states in the AFM junction of 7- and 8-ZGNRs.
Figures 6(a) and (b) show the current-voltage (-) curves for the up- and down-spin states in the FM junctions of 7- and 8-ZGNRs. It is found that the current for 7-ZGNR linearly depends on the bias voltage, while the current for 8-ZGNR is saturated at the bias voltage of about . The distinct behavior of 8-ZGNR from 7-ZGNR can be more definitely seen in the AFM junction as shown in Figs. 6(c) and (d). Interestingly, 8-ZGNR with the AFM junction exhibits a diode behavior for the spin resolved current. Only the up-spin state contributes substantially to the current in the negative bias regime. In contrary in the positive bias regime only the down-spin state contributes to the current. It is worth mentioning that the effect can also be regarded as a spin filter effect. On the other hand, the - characteristics for 7-ZGNR is nearly equivalent to that for the FM junction. The considerable difference between 7- and 8-ZGNRs in the - characteristics can be attributed to the symmetry of two wave functions, and states, around the Fermi level. For 8-ZGNR the wave functions of the and states are antisymmetric and symmetric with respect to the mirror plane which is the mid plane between two edges, respectively, while those wave functions for 7-ZGNR are neither symmetric nor antisymmetric. From a detailed analysis[80], it can be concluded that the unique distinction in the - characteristics arises from an interplay between the symmetry of wave functions and band structures of ZGNRs. In addition, we find that spin moments are reduced by applying the finite bias voltage as shown in Fig. 7. Since the flat bands around X-point of the minority spin state are located about 0.25 eV above the Fermi level, the spin moments at the zigzag edges are largely reduced by increasing the occupation of the flat bands for the minority spin states when the bias voltage exceeds the threshold as shown in Fig. 7.[78, 79] The details of the analysis on the unique spin diode and filter effect of ZGNRs is discussed elsewhere.[80]
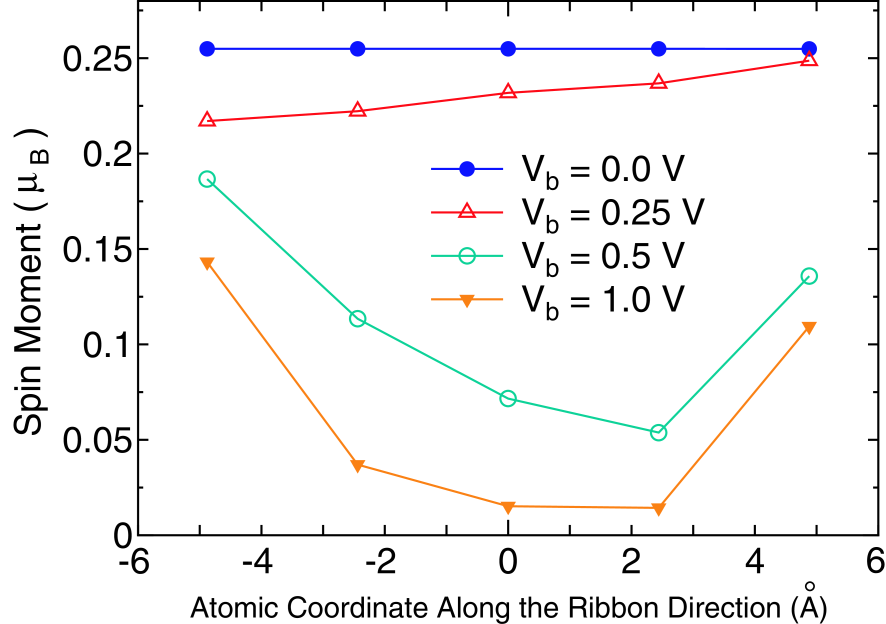
Figure 7: (Color online) Spin moments of the edge carbons in the central region for the FM junction of 8-ZGNR under various applied source-drain bias voltages .
4.4.2 Fe/MgO/Fe tunneling junction
The applicability of our implementation to bulk systems is demonstrated by an application to a tunneling junction consisting of MgO(100) layers sandwiched by iron. The magneto-tunneling junction was theoretically predicted to exhibit a large tunneling-magnetoresistance (TMR),[81] and subsequently the TMR effect has been experimentally confirmed.[82] We consider four MgO(100) layers sandwiched by iron with the (100) surface of which atomic configuration is shown in Fig. 8(a), where the lattice constant of the bc-plane used is 2.866 Å, and they are 2.866 and 4.054 Å in iron and MgO regions along the a-axis, and the distance between the MgO and iron layers is assumed to be 2.160 Å. The four MgO(100) layers corresponds to the region in Fig. 1(a), and four Fe layers of the left and right hand correspond the region and , respectively. The SCF calculations were performed at 1000 K under zero bias voltage using k-points of and 130 poles for the integration of the equilibrium density matrix. It is found that obtaining the SCF is much harder than the case of ZGNR discussed before, and that a careful and modest treatment for the charge mixing is required.
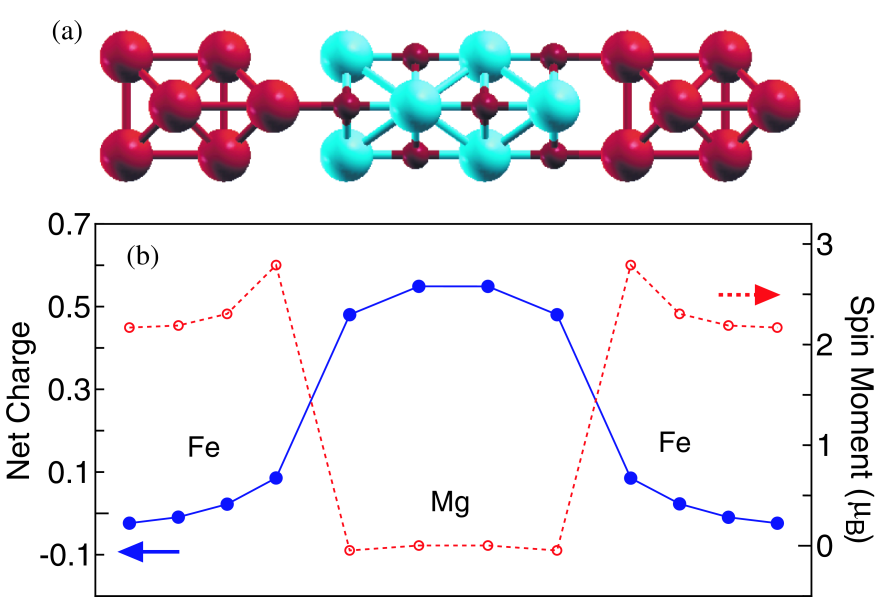
Figure 8: (Color online) (a) Calculation model for a tunneling junction consisting of four MgO(100) layers, being the region, sandwiched by Fe(100), being the and regions. The red large and small and blue circles denote Fe, O, and Mg atoms, respectively. (b) The net charge and spin magnetic moment of a Fe or Mg atom belonging to each layer in the parallel magnetic configuration between the left and right leads. The position in the horizontal axis exactly corresponds to that of the layer in the above figure.
As shown in Fig. 8(b), the net charge of iron atoms in the interfacial layer is positive due to the coordination to an oxygen atom, and the reduction of electrons leads to the increase of the magnetic moment of iron atoms at the interfacial layer. The increase of the magnetic moment at the interfacial layer makes the distinction of the majority and minority spin states at the Fermi energy clear, i.e., the nature of the majority and minority spin states at the Fermi energy can be assigned to - and -states, respectively. The k-resolved transmissions at the chemical potential for the majority and minority spin states for the parallel magnetic configuration between the left and right leads are shown in Figs. 9(a) and (b), respectively. The large peak at the point in the majority spin state can be attributed to the -state, while sharp peaks around four pillars come from the -state as discussed in Ref.[81]. Note that the position of the sharp peaks is rotated by 45 degrees because of the unit cell rotated by 45 degrees compared to that in Ref.[81]. The conductances, and , for the majority and minority spin states, calculated from the average transmission integrated over the first Brillouin zone, are 11.99 and 2.82 , respectively, which implies that the tunneling junction may behave as a spin filter. The distinction in the conductance should be attributed to decay properties of states in the insulating MgO region coupled with the two states.[81] For the antiparallel magnetic configuration the k-resolved transmission at the chemical potential is understood as a multiplication of the transmissions for the majority and minority spin states in the parallel configuration as shown in Fig. 9(c). The conductance, , of the antiparallel magnetic configuration is 0.34 () which is smaller than those of the parallel case. By defining TMR, we obtain TMR of 2082 %, which is compared to 3700 % for a 5 layer MgO case reported in Ref.[83].
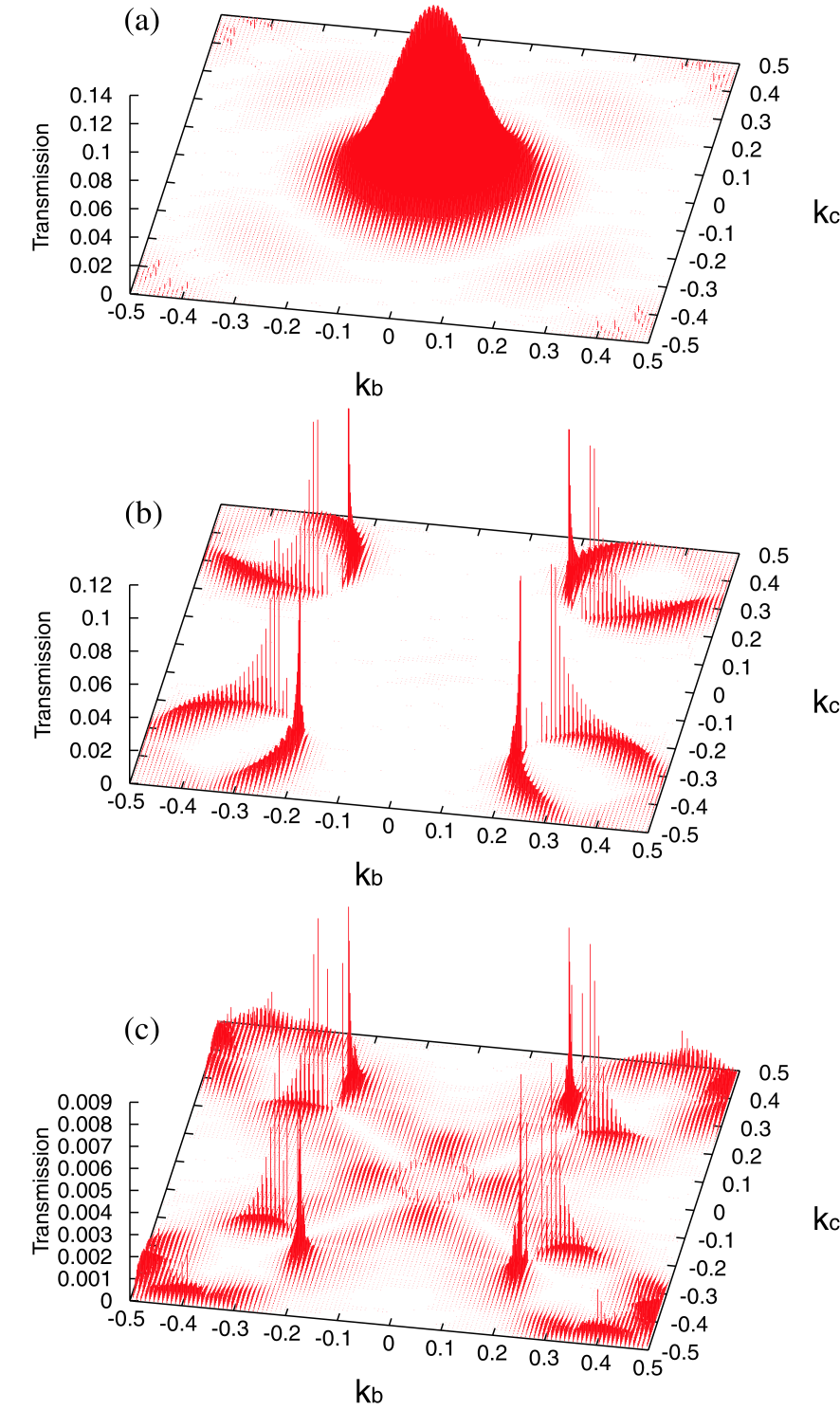
Figure 9: (Color online) k-resolved Transmission at the chemical potential for (a) the majority spin state of the parallel configuration, (b) the minority spin state of the parallel configuration, and (c) a spin state of the antiparallel configuration, respectively. For the calculations k-points of were used.
4.4.3 LaMnOSrMnO superlattice
When the transmission of a system with the periodicity along the a-axis as well as the periodicity of the bc-plane is evaluated under zero bias voltage, it can be easily obtained by making use of the Hamiltonian calculated by the conventional band structure calculation without employing the Green function method described in the paper. This scheme enables us to explore transport properties for a wide variety of possible geometric and magnetic structures with a low computational cost, and thereby can be very useful for many materials such as supperlattice structures. Once the Hamiltonian and overlap matrices are obtained from the conventional band structure calculation for the periodic structure, the transmission is evaluated by Eq. (51), where all the necessary information to evaluate Eq. (51) can be reconstructed by the result of the band structure calculation. As an example of the scheme, we calculate the conductance of a (LaMnO)/(SrMnO) superlattice with four different magnetic structures, i.e., ferromagnetic, A-type, G-type, and C-type antiferromagnetic configurations of Mn sites.[84, 85]
In recent years, it has been found experimentally that the superlattice structures consisting of LaMnO and SrMnO layers exhibit a metal-insulator transition in terms of the layer thickness.[84] Bhattacharya et al. fabricated (LaMnO)/(SrMnO) superlattices on a SrTiO (001) substrate, and measured the in-plane resistivity as a function of temperature.[84] The resistivity measurement indicates that the superlattices are metallic and insulating for and , respectively. On the other hand, the (LaMnO)/(SrMnO) superlattices fabricated on the same substrate by Nakano et al. exhibit a sample dependence in the resistivity measurement, i.e., one of three samples is metallic and the others are insulating.[85] They argued that the metallic behavior observed in the one sample may be attributed to a certain structural incompleteness in the superlattice structure, and that the ideal superlattice should become insulating based on their experimental results. A theoretical model calculation for the (LaMnO)/(SrMnO) superlattices suggests that the metal-insulator transition at can be explained by existence of the G-type antiferromagnetic barrier in the SrMnO layers sandwiched by the LaMnO layers with the ferromagnetic configuration.[86] Since the analysis by Nakano et al. suggests that the charge transfer between the LaMnO and SrMnO layers is rather localized in the vicinity of the interface,[85] the model should be applicable to the case of (LaMnO)/(SrMnO) without significantly depending on the ratio between the thicknesses of (LaMnO) and (SrMnO) layers, indicating that (LaMnO)/(SrMnO) is metallic. However, the naive consideration evidently contradicts the experimental result.[85]
As a first step towards comprehensive understanding of transport properties of the superlattice structures by the first principle calculations, we consider the simplest superlattice, i.e., (LaMnOSrMnO). In the calculations, the in-plane lattice constant is fixed to be 3.905 Å which is equivalent to that of the SrTiO substrate. The out-of-plane lattice constant is assumed to be 7.735 Å, since those are experimentally determined to be 3.959 Å and 3.776 Å for the LaMnO and the SrMnO layers, respectively, grown on the SrTiO substrate and the average out-of-plane lattice constant for the superlattices is nearly equivalent to the average of the two values.[85] With those lattice constants internal structural parameters are optimized for each magnetic configuration without any constraint until the maximum force is less than hartree/bohr. The optimized structure for the ferromagnetic configuration is shown in Fig. 10(a). It is found that the position of oxygen atoms is largely distorted due to the different ionic radii between La and Sr atoms, showing that Mn atoms are located in the center of each distorted octahedron. Also, bond angles of Mn-O-Mn are found to be 167.4 and 161.6 (degree) for the in-plane and out-of-plane, respectively. The total energies relative to the ferromagnetic configuration are listed in Table 1. The calculated ground state is the ferromagnetic configuration, and the A-type antiferromagnetic configuration lies just above 5 meV per formula unit. It may be considered that the nearly degeneracy between the two configurations corresponds to the neighborhood of the boundary at and in the phase diagram for the tetragonal LaSrMnO.[87] The two configurations have both a metallic DOS, while the ferromagnetic configuration is half-metallic as shown in Figs. 10(b) and (c), reflecting the large in-plane and out-of-plane conductances as shown in Table 1. From a detailed analysis (not shown) of DOSs, we see that the electronic states at the Fermi level are composed of orbitals of Mn atoms and orbitals of oxygen atoms. Also, the Mulliken population analysis (not shown) suggests that the charge state of Mn atoms in the superlattice is in between those in the LaMnO and SrMnO bulks. This implies that the metallic band structures are induced by partial filling of the bands. Since the bond angle of Mn-O-Mn for the out-of-plane is slightly acute, therefore, this may be attributed to reduction of the conductance of the ferromagnetic state in the out-of-plane as shown in Table 1. A systematic study for the thicker cases and the effect of Coulomb interaction[88] in the orbitals is highly desirable, and the details will be discussed elsewhere.
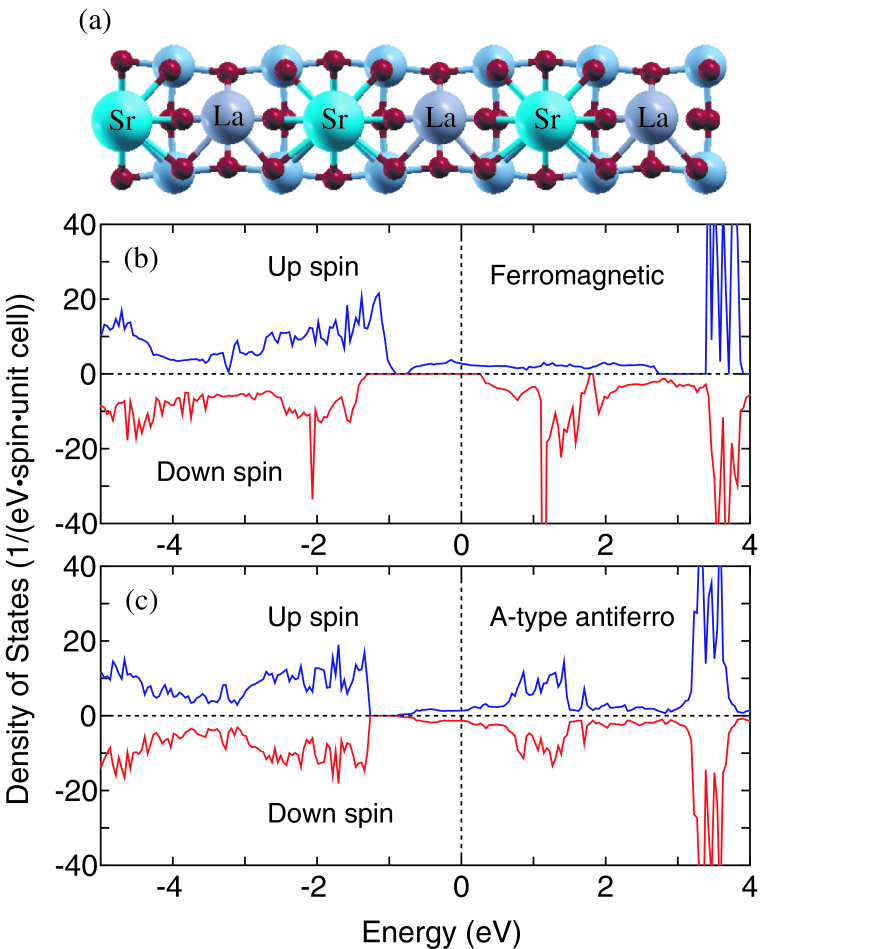
Figure 10: (Color online) (a) Optimized geometric structure of (LaMnO)/(SrMnO) superlattice with the ferromagnetic spin configuration of Mn sites. The middle sized blue and small red circles denote Mn and O atoms, while Sr and La atoms are denoted by the mark. Density of states (DOS) of the (LaMnO)/(SrMnO) superlattice for (b) ferromagnetic and (c) A-type antiferromagnetic configurations.
| F | A | C | G | |
|---|---|---|---|---|
| Energy | 0 | 5.0 | 163.8 | 248.2 |
| 2262 | 1433 | 1169 | 1646 | |
| 1425 | 1105 | 1646 | ||
| 1741 | 664 | 1127 | 678 | |
| 655 | 1128 | 677 |
4.5 Parallelization
The computation in the the NEGF method can be parallelized in many aspects such as k-points, energies in the complex plane at which the Green functions are evaluated, spin index, matrix multiplications, and calculation of the inverse of the matrix. Here we demonstrate a good scalability of the NEGF in the parallel computation by a hybrid scheme using the message passing interface (MPI) and OpenMP which are used for inter-nodes and intra-node parallelization, respectively. The Green function defined by Eq. (6) is specified by the k-point, energy , and spin index . Since the calculation of the Green function specified by each set of three indices can be independently performed without any communication among the nodes, we parallelize triple loops corresponding to the three indices using MPI. Each node only has to calculate the Green functions for an allocated domain of the set of indices, and partly sum up Eq. (26) or (44) in a discretized form. After all the calculations finish, a global summation among the nodes is required to complete the calculation of Eq. (26) or (44), which, in most cases, is a very small fraction of the computational time even including the MPI communication among the nodes. Thus, the reduction of scalability for the parallelization of the three indices is mainly due to imbalance in the allocation of the domain of the set of indices. The imbalance can happen in the case that the number of combination for the three indices and the number of processes in the MPI parallelization are relatively small and large, respectively. In addition to the three indices, one may notice that the matrix multiplications and the calculation of the matrix inverse can be parallelized, which are situated in the inner loops of the three indices. The evaluation of the central Green function given by Eq. (6) and the surface Green functions given by Eq. (21), and the self energies given by Eqs. (7) and (8) are mainly performed by the matrix multiplications and the calculation of the matrix inverse. We parallelize these two computations using OpenMP in one node. Since the memory is shared by threads in the node, the communication of the data is not required unlike the MPI parallelization. However, the conflict in the data access to the memory can reduce the scalability in the OpenMP parallelization. As a whole, in our implementation the k-point, energy , and spin index are parallelized by MPI, and the matrix multiplications and the calculation of the matrix inverse are parallelized by OpenMP.
In Fig. 11 we show the speed-up ratio in the elapsed time for the evaluation of the density matrix of 8-ZGNR under a finite bias voltage of 0.5 eV. The geometric and magnetic structures and computational conditions for 8-ZGNR are same as before. The energy points of 197 (101 and 96 for the equilibrium and nonequilibrium terms, respectively) are used for the evaluation of the density matrix. Only the point is employed for the k-point sampling, and the spin polarized calculation is performed. Thus, the combination of 394 for the three indices are parallelized by MPI. It is found that the speed-up ratio of the flat MPI parallelization, corresponding to 1 thread, reasonably scales up to 64 processes. Furthermore, it can be seen that the hybrid parallelization, corresponding to 2 and 4 threads, largely improves the speed-up ratio. By fully using 64 quad core processors, corresponding to 64 processes and 4 threads, the speed-up ratio is about 140, demonstrating the good scalability of the NEGF method.
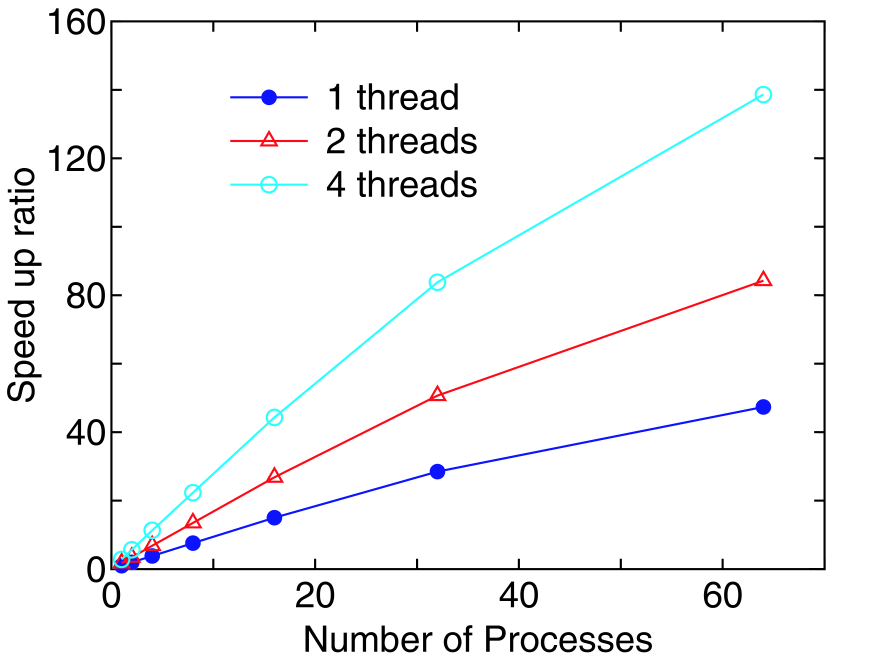
Figure 11: (Color online) Speed-up ratio in the parallel computation of the calculation of the density matrix for the FM junction of 8-ZGNR by a hybrid scheme using MPI and OpenMP. The speed-up ratio is defined by , where and are the elapsed times by a single core and a parallel calculations. The cores used in the MPI and OpenMP parallelizations are called process and thread, respectively. The parallel calculations were performed on a Cray XT5 machine consisting of AMD opteron quad core processors (2.3GHz). In the benchmark calculations, the number of processes is taken to be equivalent to that of processors. Therefore, in the parallelization using 1 or 2 threads, 3 or 2 cores are idle in a quad core processor.
5 CONCLUSIONS
We have presented an efficient and accurate implementation of the NEGF method for electronic transport calculations in combination with DFT using pseudo-atomic orbitals and pseudopotentials. In the implementation, we have developed accurate methods for the evaluation of the density matrix and the treatment of the boundary between the scattering region and the leads.
A contour integration method with a continued fraction representation of the Fermi-Dirac function has been successfully applied for the evaluation of the equilibrium term in the density matrix, which evidently outperforms the previous method,[18] while a simple quadrature scheme on the real axis with a small imaginary part is employed for that for the nonequilibrium term in the density matrix. It has been demonstrated by numerical calculations that the accuracy of eV per atom in is attainable using the energy points of 200 in the complex plane even under a finite bias voltage of 0.5 V at 600 K. However, the evaluation of the nonequilibrium density matrix still requires a careful treatment where the pole structure of the Green functions has to be smeared by introducing a finite imaginary part. The numerical calculations suggest that the number of energy points required for the convergence can be largely reduced by introducing the imaginary part of 0.01 eV without largely changing the calculated results in a practical sense. We also note that the accurate evaluation of the density matrix provides another advantage that the SCF calculations even under a finite bias voltage smoothly converge in a similar fashion as the conventional band structure calculation does.
We have also developed an efficient method for calculating the Hartree potential by a combination of the two dimensional FFT and a finite difference method without any ambiguity in reproducing the boundary conditions. In addition, a careful evaluation of the charge density near the boundary between the scattering region and the leads is presented in order to avoid the spurious scattering accompanied by the inaccurate construction of the charge density. The proper treatment for the charge evaluation in our implementation can definitely be verified by a comparison between the conventional band structure calculation and the EGF method with respect to the DOS of the carbon chain.
Finally, we have demonstrated the applicability of our implementation by calculations of spin resolved - characteristics of ZGNRs, showing that the - characteristics depend on the symmetry of ZGNR, and that the symmetric ZGNR exhibits a unique spin diode and filter effect. Also, the applicability of our implementation to bulk systems is demonstrated by applications to a Fe/MgO/Fe tunneling junction and a LaMnOSrMnO superlattice. Based on the above discussions and the good parallel efficiency in the hybrid parallelization shown in the study, it is concluded that our implementation of the NEGF method can be applicable to challenging problems related to large-scale systems, and can be a starting point, apart from numerical spurious effects, to include many body effects beyond the one particle picture in the electronic transport.
Acknowledgments
The authors would like to thank H. Kondo for providing a prototype FORTRAN code of the NEGF method. This work is partly supported by CREST-JST, the Next Generation Supercomputing Project, Nanoscience Program, and NEDO (as part of the Nanoelectronics project).
Appendix A: An energy functional for the equilibrium state
Let us introduce the following density functional which may define the total energy of the central region being a part of the extended system at equilibrium with a common chemical potential :
| (57) |
where is the Coulomb interaction energy between electrons and the external potential of the central region given by
| (58) |
and is the Hartree energy defined by
where is an additional electron density which arises from the boundary condition between the central and lead regions. In fact, the additional electron density can be obtained by back Fourier transforming Eq. (37) divided by within our treatment. The last term of Eq. (57), , is a self energy density functional, which may correspond to the energy contribution from the self energy due to the semi infinite leads, given by
| (60) |
with
| (61) |
where , the factor of 2 is due to the spin multiplicity, and the self energy is the sum of the self energies arising from the left and right leads. Although we neglect the spin and k-dependency on the formulation for the simplicity throughout the Appendix, its generalization with the dependency is straightforward. The kinetic energy can be evaluated by Eq. (6) as the band energy minus double counting corrections as follows:
| (62) | |||||
where is defined by
| (63) |
It is noted that the last term in Eq. (62) cancels the contribution from the first term in Eq. (61). The effective potential in the second term of Eq. (62) will be defined later. Also, the exchange-correlation energy in Eq. (57) is considered to be a density functional such as local density approximations (LDA) and generalized gradient approximations (GGA) evaluated using electron density in the central region .
We now consider the variation of the energy with respect to . The variations of and are simply given by
| (64) |
and
| (65) |
By noting the Dyson equation and which are both derived from Eq. (6), and , the variation of is given by two contributions:
The trace in the first term of Eq. (A) can be transformed by considering a partial integral and assuming the system to be insulating as follows:
| (67) | |||||
| (68) |
It is also noted that the second term in Eq. (A) cancels the variation of the contribution from the second term in Eq. (61). The variation of the second term in Eq. (62) is easily found as
| (69) |
Thus, it turns out using Eqs. (64)-(69) that the variation of the energy is given by
| (70) |
Letting be zero so that the variation of the energy can be always zero with respect to , we obtain a form of the effective potential:
| (71) |
It is found that the effective potential takes the same form as in the Kohn-Sham method.[41] The fact implies that the self consistent solution of the Green function under the zero bias condition may correspond to the minimization of the energy functional defined by Eq. (57), since in practice the Green function defined by Eq. (6) is calculated using the effective potential given by Eq. (71) as a consequence of combining the NEGF method with DFT.
The generalization of the functional to the metallic case with a finite temperature and the nonequilibrium state might be an important direction in the future study so that forces on atoms can be variationally calculated from the functional, since the existence of a variational functional has been recently suggested for the nonequilibrium steady state.[89, 90, 91]
Appendix B: Energy density matrix
The equilibrium energy density matrix , where one of the associated basis orbitals is in the central cell and the other is in the cell denoted by , is calculated using the contour integration method applied to the equilibrium density matrix as follow:
| (72) |
with
| (73) |
If the Hamiltonian and overlap matrices are -independent, as well as Eq. (22), Eq. (73) can be simplified to:
| (74) |
For the general case with the -dependent Hamiltonian and overlap matrices, Eq. (73) is evaluated by the contour integration method with the special form of Fermi-Dirac function given by Eq. (25) as follows:
with
| (76) |
where is the first order moment of the Green function . In Eq. (B), a term, , which appears mutually for , is omitted, since the diverging terms cancel each other out in Eq. (72). By making use of the moment representation of the Green function,[45] the following simultaneous linear equation is derived for the zero and first order moments:
| (77) |
Letting and be and , respectively, the zero and first order moments can be evaluated by
| (78) | |||||
| (79) |
where is a large real number so that the higher order moments can be neglected.
For the nonequilibrium Green function, the nonequilibrium contribution in the the energy density matrix can be calculated using the simple quadrature scheme in the same way as for the nonequilibrium term in the density matrix.
References
- [1] J. Schwinger, J. Math. Phys. 2, 407 (1961).
- [2] L.V. Keldysh, Soviet Phys. JETP 20, 1018 (1965).
- [3] C. Caroli, R. Combescot, P. Nozieres, and D. Saint-James, J. Phys. C 4, 916 (1971).
- [4] S. Datta, Electronic Transport in Mesoscopic Systems (Cambridge University Press, 1997).
- [5] S. Datta, Quantum Transport: Atom To Transistor, (Cambridge University Press, 2005).
- [6] H. Haug and A.-P. Jauho, Quantum Kinetics in Transport and Optics of Semiconductors, (Springer; 2nd, 2007).
- [7] P.A. Derosa and J.M. Seminario, J. Phys. Chem. B 105, 471 (2001).
- [8] H. Kondo, H. Kino, J. Nara, T. Ozaki, and T. Ohno, Phys. Rev. B 73, 235323 (2006).
- [9] Y. Wei, Y. Xu, J. Wang, and H. Guo, Phys. Rev. B 70, 193406 (2004).
- [10] A. Grigoriev, N.V. Skorodumova, S. I. Simak, G. Wendin, B. Johansson, and R. Ahuja, Phys. Rev. Lett. 97, 236807 (2006).
- [11] Z. Li, H. Qian, J. Wu, B.-L. Gu, and W. Duan, Phys. Rev. Lett. 100, 206802 (2008).
- [12] W.Y. Kim and K.S. Kim, Nature Nanotech. 100, 408 (2008).
- [13] S.-H. Chen, C.-R. Chang, J.Q. Xiao, and B.K. Nikolic, Phys. Rev. B 79, 054424 (2009).
- [14] A. Bulusu and D.G. Walker, J. Appl. Phys. 102, 073713 (2007).
- [15] M.B. Nardelli, J.-L. Fattebert, and J. Bernholc, Phys. Rev. B, 64, 245423 (2001).
- [16] P. S. Damle, A. W. Ghosh, and S. Datta, Phys. Rev. B, 64, 201403 (2001).
- [17] J. Taylor, H. Guo, and J. Wang, Phys. Rev. B 63, 245407 (2001).
- [18] M. Brandbyge, J.-L. Mozos, P. Ordejon, J. Taylor, and K. Stokbro, Phys. Rev. B 65, 165401 (2002).
- [19] F.D. Novaes, A.J.R. da Silva, A. Fazzio, Brazilian J. Phys. 36, 799 (2006).
- [20] W.Y. Kim and K.S. Kim, J. Comput. Chem. 29, 1073 (2008).
- [21] R. Li, J. Zhang, S. Hou, Z. Qian, Z. Shen, X. Zhao, and Z. Xue, Chem. Phys. 336, 127 (2007).
- [22] A.R. Rocha, V.M. Garcia-Suarez, S. Bailey, C. Lambert, J. Ferrer, and S. Sanvito, Phys. Rev. B 73, 085414 (2006).
- [23] L.-N. Zhao, X.-F. Wang, Z.-H. Yao, Z.-F. Hou, M. Yee, X. Zhou, S.-H. Lin, T.-S. Lee, J. Comput Electron 7, 500 (2008).
- [24] P. Havu, V. Havu, M. J. Puska, and R. M. Nieminen, Phys. Rev. B 69, 115325 (2004).
- [25] T. Shimazaki, H. Maruyama, Y. Asai, and K. Yamashita, J. Chem. Phys. 123, 164111 (2005).
- [26] T. Shimazaki, Y. Xue, M.A. Ratner, and K. Yamashita, J. Chem. Phys. 124, 114708 (2006).
- [27] H. Wang and G.K.-L. Chan, Phys. Rev B 76, 193310 (2007).
- [28] Y. Asai, Phys. Rev. Lett. 93, 246102 (2004).
- [29] N. Sergueev, D. Roubtsov, and H. Guo, Phys. Rev. Lett. 95, 146803 (2005).
- [30] M. Paulsson, T. Frederiksen, and M. Brandbyge, Phys. Rev B 72, 201101R (2005).
- [31] N. Sergueev, A.A. Demkov, and H. Guo, Phys. Rev. B 75, 233418 (2007).
- [32] T. Shimazaki and Y. Asai, Phys. Rev B 77, 075110 (2008).
- [33] T. Shimazaki and Y. Asai, Phys. Rev B 77, 115428 (2008).
- [34] A. Ferretti, A. Calzolari, R.D. Felice, F. Manghi, M.J. Caldas, M.B. Nardelli, and E. Molinari, Phys. Rev. Lett. 94, 116802 (2005).
- [35] P. Darancet, A. Ferretti, D. Mayou, and V. Olevano, Phys. Rev. B 75, 075102 (2007).
- [36] K. Thygesen and A. Rubio, J. Chem. Phys. 126, 091101 (2007).
- [37] K.S. Thygesen and A. Rubio, Phys. Rev. B 77, 115333 (2008).
- [38] A.R. Williams, P.J. Feibelman, and N.D. Lang, Phys. Rev. B 26, 5433 (1982).
- [39] M. Paulsson, arXiv:cond-mat/0210519v2.
- [40] P. Hohenberg and W. Kohn, Phys. Rev. 136, B 864 (1964).
- [41] W. Kohn and L. J. Sham, Phys. Rev. 140, A 1133 (1965).
- [42] T. Ozaki, Phys. Rev. B. 67, 155108 (2003).
- [43] T. Ozaki and H. Kino, Phys. Rev. B. 69, 195113 (2004).
- [44] T. Ozaki and H. Kino, J. Chem. Phys. 121, 10879 (2004).
- [45] T. Ozaki, Phys. Rev. B. 75, 035123 (2007).
- [46] M.P. Lopez Sancho, J.M. Lopez Sancho, J.M.L. Sancho, and J. Rubio, J. Phys. F 15, 851 (1985).
- [47] D.M.C. Nicholson, G.M. Stocks, Y. Wang, and W.A. Shelton, Phys. Rev. B 50, 14686 (1994).
- [48] S. Goedecker, Phys. Rev. B 48, 17573 (1993).
- [49] K. Wildberger, P. Lang, R. Zeller, and P.H. Dederichs, Phys. Rev. B 52, 11502 (1995).
- [50] V. Drchal, J. Kudrnovsky, P. Bruno, I. Turek, P.H. Dederichs, and P. Weinberger, Phys. Rev. B 60, 9588 (1999).
- [51] T. Matsubara, Prog. Theor. Phys. 14, 351 (1955).
- [52] T. Ozaki and H. Kino, Phys. Rev. B. 72, 045121 (2005).
- [53] In a similar fashion to the charge density, the atomic electron density in the central region consists of two contributions. One of them comes from atoms located in the central region , and the other is the contribution by the tail of atomic electron densities associated with atoms in the leads regions. Since the latter is independent of the the SCF iteration, it is computed on the regular mesh before the SCF iteration.
- [54] G. P. Kerker, Phys. Rev. B 23, 3082 (1981).
- [55] G. Kresse and J. Furthmeuller, Phys. Rev. B. 54, 11169 (1996).
- [56] G. C. Liang, A. W. Ghosh, M. Paulsson, and S. Datta, Phys. Rev. B. 69, 115302 (2004).
- [57] H. J. Monkhorst and J. D. Pack, Phys. Rev. B 13, 5188 (1976).
- [58] Y. Meir and N.S. Wingreen, Phys. Rev. Lett. 68, 2512 (1992).
- [59] N. Troullier and J.L. Martins, Phys. Rev. B 43, 1993 (1991).
- [60] O.F. Sankey and D.J. Niklewski, Phys. Rev. B 40, 3979 (1989).
- [61] D.M. Ceperley and B.J. Alder, Phys. Rev. Lett. 45, 566(1980); J.P. Perdew and A. Zunger, Phys. Rev. B 23, 5048 (1981).
- [62] J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996).
- [63] J.M. Soler, E. Artacho, J.D. Gale, A. Garcia, J. Junquera, P. Ordejon, and D. Sanchez-Portal, J. Phys.: Condens. Matter 14, 2745 (2002).
- [64] The code, OpenMX, pseudo-atomic basis functions, and pseudopotentials are available on a web site (http://www.openmx-square.org/).
- [65] Let us consider a finite system consisting of the central region and finite leads. Since the system is finite, the total energy is well defined, and it is apparent that the standard DFT approach is applicable to the finite system. Then, we enlarge the size of the leads in the system, while keeping the finiteness, for which the standard DFT can be still applicable. Because of the finiteness of the system the DFT approach is valid even for the system with extremely large leads. The infinite system we consider may be regarded as the limiting case of the finite system.
- [66] For example, in the contour integration method in Ref. [18], the optimal choice of parameters such as , EB, and may depend on systems.
- [67] We used eV, EB=27.21 eV, and , being the parameters in the closed contour integration depicted in Fig. 2 of Ref.[18]. The value for gives four enclosed Matsubara poles for K. The path toward the positive direction is extended by from the chemical potential. The paths and are integrated using the Gauss-Legendre quadrature scheme, respectively. It turns out that 40 grid points for the path is enough to achieve the fully convergent result within double precision. So, we used 40 grid points for the path , and only changed the number of grid points for the path in the calculation for Fig. 2(a) of Ref.[18]. Therefore, the number of energy points in the closed contour integration is given by 4 (the enclosed Matsubara poles) plus 40 (the path ) plus the number of grid points for the path .
- [68] M. Fujita, K. Wakabayashi, K. Nakada, and K. Kusakabe, J. Phys. Soc. Jpn. 65, 1920 (1996).
- [69] S. Okada and A. Oshiyama, Phys. Rev. Lett. 87, 146803 (2001).
- [70] Y.-W. Son, M.L. Cohen, and S.G. Louie, Nature 444, 347 (2006).
- [71] D.A. Abanin, P.A. Lee, and L.S. Levitov, Phys. Rev. Lett. 96, 176803 (2006).
- [72] O.V. Yazyev and M.I. Katsnelson, Phys. Rev. Lett. 100, 047209 (2008).
- [73] V. M. Karpan, G. Giovannetti, P. A. Khomyakov, M. Talanana, A. A. Starikov, M. Zwierzycki, J. van den Brink, G. Brocks, and P. J. Kelly, Phys. Rev. Lett. 99, 176602 (2007).
- [74] V. M. Karpan, P. A. Khomyakov, A. A. Starikov, G. Giovannetti, M. Zwierzycki, M. Talanana, G. Brocks, J. van den Brink, and P. J. Kelly, Phys. Rev. B 78, 195419 (2008).
- [75] M. Ezawa, Eur. Phys. J. B 67, 543 (2009).
- [76] T.B. Martins, A.J. R. da Silva, R.H. Miwa, and A. Fazzio, Nano Lett. 8, 2293 (2008).
- [77] J. Guo, D. Gunlycke, and C.T. White, Appl. Phys. Lett. 92, 163109 (2008).
- [78] D.A. Areshkin, and C.T. White, Nano Lett. 7, 3253 (2007).
- [79] D. Gunlycke, D.A. Areshkin, J. Li, J.W. Mintmire, and C.T. White, Nano Lett. 7, 3608 (2007).
- [80] T. Ozaki, K. Nishio, and H. Weng, H. Kino, arXiv:0905.2461v1.
- [81] W.H. Butler, X.-G. Zhang, and T.C. Schulthess, Phy. Rev. B 63, 054416 (2001).
- [82] S. Yuasa, T. Nagahama, A. Fukushima, Y. Suzuki, and K. Ando, Nature Mater. 3, 868 (2004).
- [83] D. Waldron, V. Timoshevskii, Y. Hu, K. Xia, and H. Guo, Phys. Rev. Lett. 97, 226802 (2006).
- [84] A. Bhattacharya, S.J. May, S.G.E. te Velthuis, M. Warusawithana, X. Zhai, B. Jiang, J.-M. Zuo, M.R. Fitzsimmons, S.D. Bader, and J.N. Eckstein, Phy. Rev. Lett. 100, 257203 (2008).
- [85] H. Nakao, J. Nishimura, Y. Murakami, A. Ohtomo, T. Fukumura, M. Kawasaki, T. Koida, Y. Wakabayashi, and H. Sawa, J. Phys. Soc. Jpn. 78, 024602 (2009).
- [86] S. Dong, R. Yu, S. Yunoki, G. Alvarez, J.-M. Liu, and E. Dagotto, Phy. Rev. B 78, 201102R (2008).
- [87] Z. Fang, I.V. Solovyev, and K. Terakura, Phy. Rev. Lett. 84, 3169 (2000).
- [88] M. J. Han, T. Ozaki, and J. Yu, Phys. Rev. B 73, 045110 (2006).
- [89] T.N. Todorov, J. Hoekstra, and A.P. Sutton, Philos. Mag. B 80, 421 (2000)
- [90] M. Di Ventra and S.T. Pantelides, Phys. Rev. B 61, 16207 (2000).
- [91] M. Di Ventra, Y.-C. Chen, and T. N. Todorov, Phys. Rev. Lett. 92, 176803 (2004).