Efficient projector expansion in LCAO
electronic structure calculations: Ver. 1.0
The original has been published
in Phys. Rev. B 72, 045121 (2005).
Abstract
A projector expansion method is presented for an efficient and accurate implementation of the first-principles electronic structure calculations using pseudo potentials and atomic basis functions. By expressing the rapidly varying local potential in the vicinity of nuclei by a separable projector expansion, the difficulty involved in the grid integration using the regular real-space grid is remarkably reduced without increasing the computational effort. To illustrate the capability, it is shown that the proposed method significantly suppresses not only a spurious oscillation in the energy curve for the atomic displacement involved in a weak interaction such as hydrogen bonding, but also the dependence of optimized structure on relative position to the real-space grid in the geometry optimization within a modest grid fineness.
I. INTRODUCTION
The electronic structure method based on a linear combination of numerical atomic local basis orbitals (LCAO)[1, 2, 3, 4, 5] and pseudo potentials [6, 7] is a possible way of extending the applicability of a density functional theory (DFT) [8] to large-scale systems, since the generalized eigenvalue problem with the resultant sparse matrices can be solved by O() methods.[9, 10, 11, 12, 13, 14] The pioneering work by Sankey et al.[1] and its generalization to a fully self-consistent field (SCF) calculation by the SIESTA group [2] enable us to deal with large-scale systems within a fully self-consistent DFT. However, in general, one of difficult problems in the implementation of these LCAO methods is how the matrix elements for the effective potential are accurately evaluated within a modest computational effort.[2, 3, 15, 16, 17, 18] Although this numerical integration for the construction of matrix elements is a highly technical issue, serious problems enough to lose the validity of calculations are often elicited when we calculate weakly interacting systems such as hydrogen bonding molecules and include deep semicore states in pseudo potentials.[2] Due to the non-integrable form of the effective potential in the conventional DFT, the numerical integration is indispensable to construct the matrix elements, even if analytic basis functions such as Gaussian functions are used. While a scheme to avoid the numerical integration is to fit the potential into integrable analytic functions,[17, 20] however, it can be regarded as just a replacement of the numerical integration with the numerical fitting.
In the LCAO methods, usually, two kinds of numerical grids are used for the numerical integration for the construction of matrix elements.[2, 3, 15, 16, 17, 18, 19] One of them is a grid decomposed into radial and angular parts such as Gauss-Legendre and Lebedev grids which are combined with a partitioning scheme to decompose a multicenter integral into one-center integrals.[3, 15, 16, 17, 18, 19] The other is a regular real-space grid similar to that used in conventional plane-wave DFT methods.[2] Although the former possesses a benefit that the total energy is independent of the rotation of system unlike the latter, the latter is superior to the former in terms of computational efficiency. Since all the basis functions use the same regular real-space grid, once the value of basis function on grid is calculated and stored in the computational memory before the SCF calculation, there is no need of the recalculation of the value of basis function on grid during the SCF calculation. Therefore, in the latter, the matrix elements are quite efficiently constructed in an element-by-element fashion for non-zero values of basis functions on grid stored in the computational memory. On the other hand, in the former, it is difficult to store the value of basis function on grid because of the requirement of the extensive memory size, since the quadrature grid is determined for each set of two atoms.
Although the regular real-space grid possesses an advantage in terms of computational efficiency, however, serious problems remain in the grid integration using the regular real-space grid, since the total energy depends on the relative position between the system and the grid. [2, 21, 22] A typical illustration is that the optimized structure depends on the initial arrangement relative to the grid position. This dependence causes a severe problem when weakly interacting systems such as hydrogen bonding molecules are calculated. In such a system a highly fine grid is needed to obtain a convergent result, which demands great computational resources. The same problem involved in the real-space grid integration is also reported in other real-space methods,[21, 22] which implies that the difficulty involved in the grid integration is a major concern in the efficient and accurate implementation of real-space methods. Thus, our aim is to establish a method which addresses the regular real-space grid for the simplicity and overcomes the difficulty involved in the grid integration within a modest computational effort. In this paper, we present a projector expansion method in which a rapidly varying local potential in the vicinity of nuclei is expressed by a separable projector expansion, and thereby the contribution of the rapidly varying local potential to matrix elements is accurately calculated in the momentum space instead of the real space. Since the real-space integration is applied to only slowly varying parts in the effective potential, as a result, the difficulty involved in the grid integration is remarkably reduced. This paper is organized as follows. In Sec. II, a projector expansion method is presented in order to overcome the difficulty involved in the grid integration in the first-principles electronic structure calculations using atomic basis functions. In Sec. III, as illustrations of the accuracy of the proposed method, it is shown that the projector expansion method significantly suppresses not only a spurious oscillation in the energy curve for the atomic displacement involved in a weak interaction such as hydrogen bonding, but also the dependence of optimized structure on relative position to the real-space grid in the geometry optimization within a modest grid fineness. In Sec. IV, we summarize the projector expansion method and the capability. Finally, in the Appendix, the analytic evaluation of force on atom in the LCAO method using the projector expansion method is briefly described.
II. PROJECTOR EXPANSION
Let us start our formulation from the total energy expression of the DFT [8] to elucidate the difficulty involved in the grid integration. In the framework of atomic basis functions ,[1, 2, 4, 5] norm-conserving pseudo potentials,[6, 7] and a local density approximation (LDA) [23] to the exchange-correlation term, the total energy in the DFT is given by the sum of the kinetic energy , the electron-core Coulomb energy , the electron-electron Coulomb energy , the exchange-correlation energy , and the core-core Coulomb energy between pseudo core charges and as follows:
| (1) |
with
| (2) |
| (3) | |||||
| (4) |
| (5) |
| (6) |
where and are the site and basis function indices, respectively. is a density matrix associated with two basis functions and , and is defined by with the LCAO coefficient , the one-particle eigenenergy , the chemical potential , and a step function . is the electron density defined by . and are the local part and non-local part in the norm-conserving pseudo potential of atom , respectively. The factor 2 in and is for the spin multiplicity. In this formulation, we consider only non spin-polarized case, non-Bloch expression of one-particle wave functions, and the LDA for simplicity, but the extensions of the below description to the spin polarized case, the Bloch expression, and a generalized gradient approximation (GGA) [24] to the exchange-correlation term are straightforward. Although the expression Eq. (1) is commonly used, it can be transformed into a more tractable form without any approximation.[1, 2] Reorganizing the sum of , and by introducing an atomic electron density , we can rewrite the sum by that of two majority contributions and consisting of short-range terms and a minority contribution consisting of a long-range term as follows:
| (7) |
with
| (8) |
| (9) |
| (10) |
where a difference electron density is defined by the difference between the electron density and the sum, , of atomic electron densities as:
| (11) | |||||
Through this paper, when the site index is dropped in the designation for a quantity specifiable with the site index, it means the inclusion of the summation over the site index except for the symbolic use. The atomic electron density is evaluated from pseudo wave functions under the confinement potential in this study.[4, 5] So, is considerably smaller than . Also, it should be noted that is finite only within the confinement radius . From the sum of the local part of pseudo potential and a Hartree potential associated with the atomic electron density, a neutral atom potential is defined by . The neutral atom potential is spherical because of the spherical atomic electron density, and becomes zero beyond the confinement radius due to Gauss’s law, indicating that it is a short-range spherical potential. Since the electron density can be evaluated the short-range quantities and , we see that the neutral atom potential energy consists of short-range terms. In addition to this, the screened core-core Coulomb energy is also evaluated by taking account of only the neighboring atoms, since the second term in Eq. (9) becomes exactly equivalent to the first term when , and thereby the long-range terms in Eq. (9) vanish. Therefore, the first and second terms being the majority contributions in the right side of Eq. (7) are calculated by making use of information of adjacent atoms in an O() operation. Since the second term in the right side of Eq. (9) is a function of only the distance between two atoms, it is accurately evaluated using a finer real-space grid, and can be tabulated as a function of the distance between two atoms at the first stage of the calculation. So, the evaluation of is far from the difficulty involved in the real-space grid integration. The difference electron-electron Coulomb energy is a minority contribution to the total energy, but is constructed by a long-range term , since the difference Hartree potential is associated with the every difference electron density in the real space. It is worth mentioning that the Ewald summation over the core-core Coulomb energy is not required by the reorganization for parts of the total energy, which is one of advantages in the reorganized expression for the total energy. Considering the variation of the total energy with respect to the LCAO coefficient , we have a well-known generalized eigenvalue equation , where and are a Hamiltonian matrix defined by and an overlap matrix defined by , respectively. In the Kohn-Sham (KS) Hamiltonian , the effective potential is given by
| (12) |
where is the exchange-correlation potential. In the matrix generalized eigenvalue equation, the matrix elements for the overlap matrix, the kinetic operator, and the non-local part of pseudo potential can be evaluated by two-center integrals. Since is expressed by a separable projector expansion,[25, 26] the matrix elements are reduced to a product of two-center integrals. The resultant two-center integrals in the evaluation of these matrix elements can be very accurately evaluated using a finer grid in the momentum space within almost the same computational time because they only have to be evaluated once before the SCF loop.[1, 2]
Rather than the matrix elements expressible by two-center integrals, the difficulty involved in the grid integration comes from the remaining contributions in the effective potential. Although the matrix elements for the remaining potentials , , and has been evaluated by the grid integration using the regular real-space grid,[2] it can be pointed out that the matrix element for the neutral atom potential is difficult to be accurately calculated within a modest fineness of the real-space grid. In Fig. 1 shows potentials , , and in Eq. (12) along the bond axis in a carbon monoxide (CO). We can see that the neutral atom potential rapidly varies in the vicinity of nuclei, while the other potentials and smoothly vary. The comparison suggests that a finer grid is required to accurately evaluate the matrix elements for the neutral atom potential , and that the calculations are not converged when a modest fineness of the real-space grid is used as shown later on.
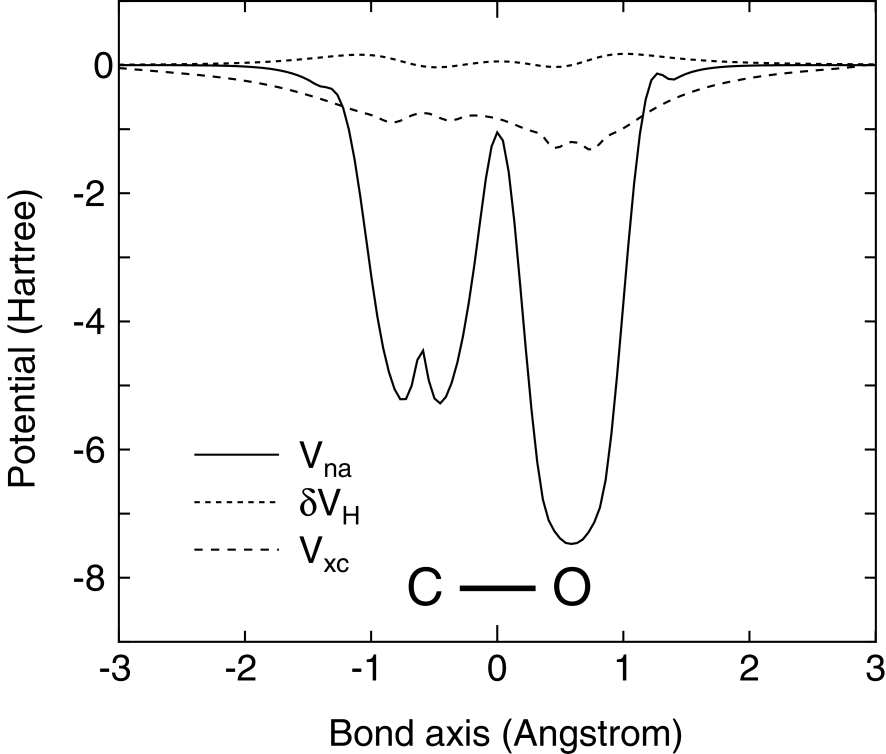
Thus, in order to suppress the difficulty involved in the real-space grid integration, we propose a projector expansion method to accurately evaluate the matrix elements for the neutral atom potential . The neutral atom potential is spherical and is defined within the finite range determined by the cutoff radius of the confinement potential. Therefore, the potential can be expressed by a projector expansion as follows:
| (13) |
where a set of radial functions is an orthonormal set defined by a norm for radial functions and , and is calculated by the following Gram-Schmidt orthogonalization:[26]
| (14) | |||||
| (15) |
The radial function used in this study is pseudo wave functions for both the ground and excited states under the same confinement potential as used in the calculation of the atomic electron density .[4, 5] It is expected that the projector expansion rapidly converges with respect to the summation over , since radial functions forms an orthonormal set within the finite range of . The projector expansion for the neutral atom potential becomes exact when the summation over the angular and the radial parts goes to infinity. The most important feature in the projector expansion defined by Eq. (13) is that the neutral atom potential is expressed by a separable form, and thereby we only have to evaluate the two-center integrals to construct the matrix elements for the neutral atom potential once before the SCF loop as well as the other two-center integrals. As discussed above, the two-center integrals can be accurately evaluated in the momentum space instead of the real space. Therefore, we can avoid the grid integration using the real-space grid for the rapidly varying neutral atom potential in the vicinity of nuclei, indicating that the difficulty involved in the grid integration is almost suppressed. It should be noted that the computational time by the introduction of the projector expansion is not increased within the SCF loop, since the basis functions and the projectors in Eq. (13) are Fourier-transformed once at the first stage of calculations, and the two-center integrals between them are evaluated before the SCF loop. Furthermore, the computational demand before the SCF loop is not large. In fact, the comparison of the total computational time shows that the computational time by the projector expansion method is only 13 % longer than that by the conventional method with the same real-space grid fineness as used in the projector expansion method in a molecular dynamics (MD) simulation (100 MD steps) of a C molecule using 4 processors of Pentium 4. In addition to this, these calculations can be easily parallelized on a parallel computer due to no data-communication in their parallel computation.
There exists another advantage of the projector expansion method. The choice of the local part in the separable pseudo potential is an important factor to enhance the transferability of the pseudo potential. However, choosing a deep local part has tended to be avoided because finer real-space mesh is necessary to calculate converged results in the real-space grid integration. In this projector expansion method, there is no difficulty in the choice of the deep local part. Thus, it is possible not only to choose the deep local part, but also to include deep semicore states in the pseudo potential without increasing computational effort.
If the same radial functions introduced in Eq. (14) are used for the basis functions () in the LCAO method, the matrix elements for the neutral atom potential are evaluated in case of and/or without depending on the convergence of the summation over the angular and radial parts. Since , e.g., in case of , we have
| (16) | |||||
Therefore, the one-center integral () and the two-center integral ( or ) are highly accurately evaluated in the evaluation of , where is referred to as integral to distinguish it from one- and two-center integrals in the projector expansion defined by Eq. (13).
Although we are able to complete our formulation for the projector expansion here, a different scheme is furthermore introduced to accurately calculate the two-center integral (). In this case, the neutral atom potential and the product of and are Fourier-transformed at the first stage of the calculation, and therefore the two-center integral between the neutral atom potential and the product of two basis functions is accurately evaluated in the momentum space apart from the projector expansion by Eq. (13). As a result, all the one- and two-center integrals, which are large components in the matrix elements, are calculated with a high degree of precision without depending on the convergence of the summation over the angular and radial parts. The projector expansion by Eq. (13) is applied only for the three-center integrals which are small components in the matrix elements. Thus, the error associated with the convergence of the summation in the projector expansion is significantly suppressed.
There is another origin of the difficulty involved in the real-space grid integration. The partial core correction (PCC) charge density is often used in Eq. (5) to take into account the non-linearity in the exchange-correlation terms. In this case, if a highly localized and large PCC charge density in the vicinity of nuclei is employed, the difficulty associated with Eq. (5) appears in the real-space grid integration. However, the difficulty can be avoidable by using a modest PCC charge density. In addition, we can avoid the inclusion of the large PCC charge density by using the pseudo potential including the semicore states, since it is relatively easy to include the semicore states in the projector expansion method as discussed above.
Here, we would like to comment on the evaluation of force on atom in the LCAO method using the projector expansion scheme. The majority contributions to the total energy are expressed by the two-center integrals in the projector expansion method. So, the contribution to the force associated with these parts can be analytically calculated by differentiating the two-center integrals evaluated in the momentum space without any difficulty. On the other hand, the remaining and are evaluated using the real-space grid integration. Consequently, it is not trivial whether the force associated with and is analytically appraisable or not. However, in fact, it is easy to analytically evaluate the force on atom in the regular real-space scheme coupled with the projector expansion method. The details are given in the Appendix.
III. NUMERICAL RESULTS
In this section, we show numerical results on the convergence of the summation over the angular and radial parts in Eq. (13) and demonstrate the capability of the projector expansion method for the suppression of the difficulty involved in the real-space grid integration.
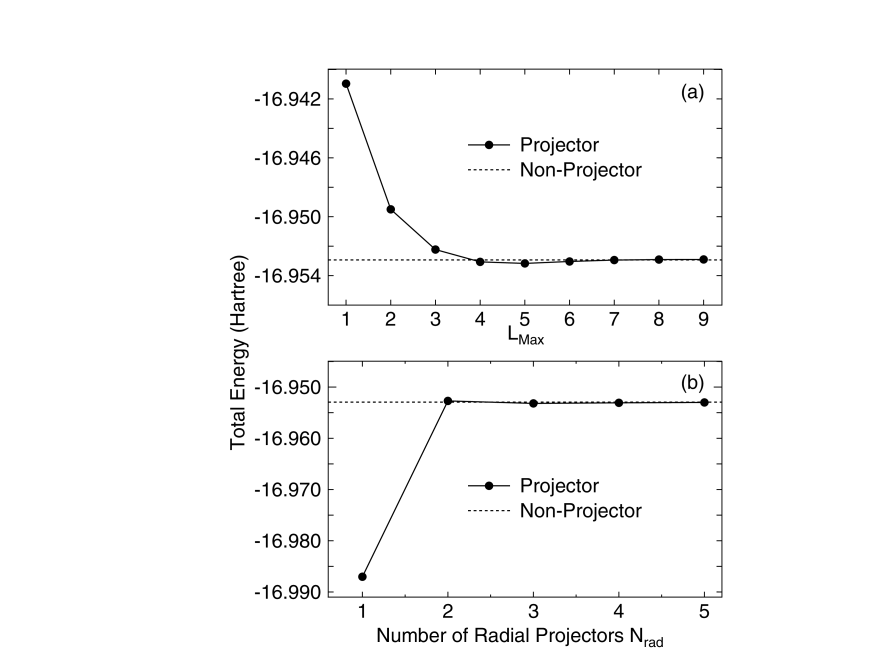
In Fig. 2 shows the convergence properties of the total energy in a water molecule as a function of the max , , and the number of radial projectors, , in the summation in Eq. (13). In this study, an optimized double valence plus polarization function (DVP) was used as a basis set.[4, 5] To replace the deep core potential into a shallow potential, a norm-conserving pseudo potential [7] was used in a separable form with multiple projectors.[26] The cutoff radii of pseudo potentials are listed in Table I of Ref.[5]. A generalized gradient approximation (GGA) [24] is used for the exchange-correlation without the nonlinear partial core correction. The cutoff energy of 3600 (Ryd) and the Gauss-Legendre grid [27] of 128 were used for the evaluation of the two-center integrals (integrals) in the momentum space. The cutoff energy for the real-space grid integration is given in the caption of the Figures and Table. All calculations were performed by our DFT code, OpenMX.[28] From the convergence properties, we see that the fully convergent result is achieved when and approach 6 and 4, respectively. In this case, we have to use at least 2 for and to accurately evaluate the one- and two-center integrals, since the double valence plus polarization function is used as a basis set. If the values are less than 2, the accuracy of the one- and two-center integrals is not assured. Based on the convergence properties, we generally use the following values: and , where is the maximum angular momentum quantum number of basis functions. From several test calculations, we confirm that the values for and are enough to achieve the fully convergent results for systems including other elements, while the results are not shown.
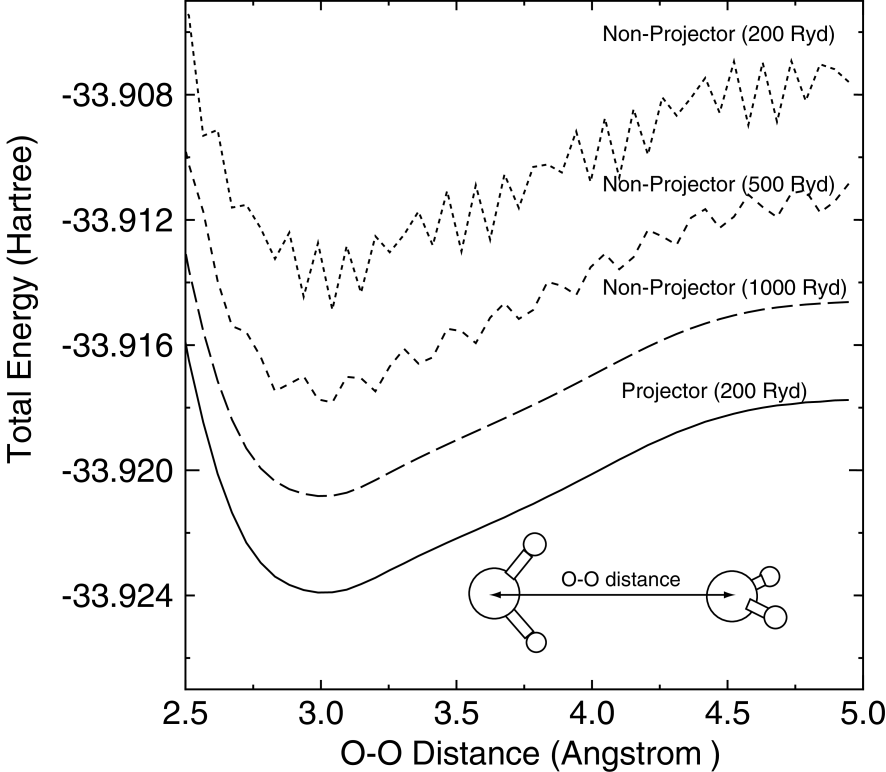
To illustrate the accuracy of the projector expansion method, we show a comparison of the total energies of a water dimer molecule, which is a typical hydrogen bonding system with a weak interaction, calculated by the projector expansion method (Projector) and the grid integration using the regular real-space grid (Non-Projector) in Fig. 3. The total energies are calculated as a function of the distance between two oxygen atoms in water molecules, each of which possesses the experimental geometry. The dihedral angle between the water molecules is fixed at 180 degrees as depicted in Fig. 3. We see that the total energy calculated by the grid integration using the regular real-space grid with a lower cutoff energy considerably oscillates, while the oscillation can be suppressed using the high cutoff energy of 1000 Ryd. This spurious oscillation is due to the inaccurate calculation of the matrix elements for the neutral atom potential around the tail of basis functions in the real-space grid integration. On the other hand, the projector expansion method entirely suppresses the spurious oscillation of the total energy curve even in the lowest cutoff energy, thus indicating that the contribution around the tail of basis functions to the matrix elements associated with the neutral atom potential are accurately calculated. The calculated energy curve by the projector method is completely consistent with that by the non-projector method using 1000 Ryd. It should be noted that the error in the calculation of the total charge is only 1.0e-5 even in the non-projector method using 200 Ryd. Nevertheless, the spurious oscillation appears in amplitude of 0.001 Hartree in the non-projector method. Our analysis clearly shows that the spurious oscillation comes from the deep neutral atom potential , and is removed by replacing it by the projector expansion.
As shown above, the spurious oscillation appears in the energy curve calculated by the real-space grid integration. Therefore, it would be considered that the optimized structure is trapped in a local minimum near the initial geometry if the geometry optimization is performed using the real-space grid integration, which could be a serious problem in the calculations of weakly interacting systems. In contrast, the difficulty is overcome in the projector expansion method. To study this dependence of the optimized structure on the initial geometry, we optimize the structure of a water molecule with various initial positions relative to the regular real-space grid. The optimized structure by the real-space grid integration strongly depends on the initial geometry relative to the regular real-space grid as shown in Table I, which indicates that the structure is trapped in a local minimum near the initial geometry. On the other hand, the optimized structure by the projector expansion method is almost independent of the initial geometry relative to the regular real-space grid, while the difference in the bond angle is 0.4 degrees at a maximum. These two clearly illustrate that the difficulty involved in the grid integration is successfully suppressed by introducing the projector expansion for the rapidly varying neutral atom potential in the vicinity of nuclei.
TABLE. I. Dependence of optimized structure (Å and degrees) and dipole moment (Debye) of a water molecule on the initial position relative to the regular real-space grid. The initial Cartesian coordinates (Å) are given by (a) O=(0.0,0.0,0.0), H=(0.76,0.59,0.0), H=(-0.76,0.59,0.0). The other initial Cartesian coordinates are generated by rotating the coordinates (a) by (b) , (c) , and (d) , where means a rotational matrix which rotates the coordinate by 30 degrees on the x-axis. The cutoff energy of 177 (Ryd) was used for the grid integration using the regular real-space grid. The experimental values are taken from Ref. [29, 30].
| Projector | |||
|---|---|---|---|
| Initial Geo. | |||
| a | 0.990, 0.990 | 104.8 | 1.95 |
| b | 0.990, 0.990 | 104.5 | 1.95 |
| c | 0.990, 0.990 | 104.8 | 1.94 |
| d | 0.990, 0.990 | 104.9 | 1.95 |
| Non-Projector | |||
| Initial Geo. | |||
| a | 0.989, 0.989 | 101.2 | 2.00 |
| b | 0.982, 0.982 | 102.0 | 1.98 |
| c | 0.993, 0.993 | 101.4 | 1.99 |
| d | 1.004, 0.993 | 107.0 | 1.92 |
| Expt. | 0.957 | 104.5 | 1.86 |
IV. CONCLUSIONS
In the real-space implementation of the DFT using atomic basis functions, one of difficult problems is how the matrix elements for the effective potential are accurately evaluated in an efficient numerical scheme within a modest computation effort. Especially, this problem becomes obvious when weakly interacting systems such as hydrogen bonding molecules are calculated. In this context, to suppress the difficulty involved in the regular real-space grid integration, we have developed a projector expansion method which expresses the rapidly varying neutral atom potential in the vicinity of nuclei by a separable projector. The neutral atom potential being the origin of the difficulty involved in the real-space grid integration is factorized by the separable projectors, and the resultant two-center integrals are accurately evaluated in the momentum space instead of the real space. To illustrate the accuracy of the projector expansion method, we have shown that the spurious oscillation in the energy curve of a water dimer molecule is entirely suppressed and that the optimized structure is almost independent of the initial geometry relative to the regular real-space grid within a modest grid fineness. Thus, we conclude that the projector expansion method is a highly useful technique for the accurate and efficient implementation of the DFT based on the LCAO method.
ACKNOWLEDGMENT
This work was partly supported by NEDO under the Nanotechnology Materials Program, Research and Development for Applying Advanced Computational Science and Technology of Japan Science and Technology Corporation (ACT-JST), and NAREGI Nanoscience Project and Special Coordination Funds of the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Appendix A
In this Appendix, we show an explicit expression for the analytic evaluation of the force on atom in the projector expansion method. On the parts of the total energy expressed by the two-center integrals (integrals), its contributions to the force are analytically calculated by differentiating the two-center integrals (integrals) evaluated in the momentum space. Then, each term in the summation over the discretized radial grid in the momentum space are analytically differentiating using cubic splines without any difficulty. Therefore, we focus on the analytic differentiation of the remaining and with respect to atomic coordinate. So, this derivation is a general formulation for the analytic force evaluation in the LCAO method based on the grid integration using the regular real-space grid. The difference electron-electron Coulomb energy and the exchange-correlation energy can be discretized using the regular real-space grid as follows:
| (17) |
| (18) |
where is the index in a vector form for specifying the position in the regular real-space grid, and is the volume per grid. Then, the derivative of with respect to the atomic coordinate is given by
| (19) |
In this study, is defined through the fast Fourier transform (FFT) of by
| (20) | |||||
where is the number of grid points in the regular real-space grid. Considering this expression of , we obtain an explicit compact expression for the derivative of with respect to as follows:
| (21) | |||||
Similarly, the derivative of with respect to is given by
| (22) | |||||
The derivative of with respect to is found by considering the definition of as follows:
| (23) | |||||
The derivative of the LCAO coefficient with respect to can be transformed to the derivative of the overlap matrix by taking account of the orthonormality relation in one-particle wave functions as usually made in the LCAO method with the non-orthogonal basis set.[1] Since the overlap matrix element is a two-center integral, the matrix element and its derivatives are evaluated in the momentum space as well as the other two-center integrals. Also, the derivative of the basis function with respect to is analytically evaluated using cubic splines because of the use of numerical basis function. The derivative of with respect to is formed by only the contribution from the same atomic site due to the independent grid position from the atomic positions as follows:
| (24) |
The derivative of with respect to the atomic coordinate is easily evaluated by
| (25) | |||||
Even for the GGA, the derivative of with respect to the atomic coordinate is explicitly expressed, while the GGA is implemented by a finite difference scheme in our implementation. As a result, the derivative of the discretized and are analytically evaluated. In addition, considering the localized basis functions , the evaluation of Eqs. (19) and (25) can be quite efficiently performed in an O() operation as well as the other contributions to the force.
The force on atom in the LCAO methods based on the numerical grid integration has been often evaluated by appealing the Hellman-Feymann theorem with the Pulay correction [3] rather than strictly differentiating the discretized energy expression. However, our derivation shows that it is possible to evaluate the analytic force consistent with the total energy in an efficient way, even though both the numerical basis functions and the numerical grid integration are introduced. A defect of the analytic forces derived here is that they do not fulfill the law of action and reaction. The defect comes from the independent determination of the regular real-space grid from the atomic positions. Since the grid position is fixed in the absolute Cartesian coordinate, the calculated force suffers from ’a pinning effect’. Nevertheless, the effect is relatively small since only the minor contributions and to the total energy are evaluated using the real-space grid integration. A way of recovering the law of action and reaction is to introduce an adaptive coordinate [31] which is dependent on the atomic positions, while the simplicity in the regular real-space grid is lost. A study toward this direction will be a future work.
References
- [1] O. F. Sankey and D. J. Niklewski, Phys. Rev. B 40, 3979 (1989).
- [2] P. Ordejon, E. Artacho, J. M. Soler, Phys. Rev. B 53, R10441 (1996); J. M. Soler, E. Artacho, J. D. Gale, A. Garcia, J. Junquera, P. Ordejon, and D. Sanchez-Portal, J. Phys.:Condens. Matter 14, 2745 (2002) and references therein.
- [3] B. Delly, J. Chem. Phys. 92, 508 (1990).
- [4] T. Ozaki, Phys. Rev. B 67, 155108 (2003).
- [5] T. Ozaki and H. Kino, Phys. Rev. B 69, 195113 (2004).
- [6] G. B. Bachelet, D. R. Hamann, and M. Schluter, Phys. Rev. B 26, 4199 (1982).
- [7] N. Troullier and J. L. Martins, Phys. Rev. B 43, 1993 (1991).
- [8] P. Hohenberg and W. Kohn, Phys. Rev. 136, B864 (1964); W. Kohn and L. J. Sham, Phys. Rev. 140, A1133(1965)
- [9] W. Yang, Phys. Rev. Lett. 66, 1438 (1991).
- [10] S. Goedecker and L. Colombo, Phys. Rev. Lett. 73, 122 (1994).
- [11] G. Galli and M. Parrinello, Phys. Rev. Lett. 69, 3547 (1992).
- [12] X.-P. Li, R. W. Nunes, and D. Vanderbilt, Phys. Rev. B 47, 10891 (1993); M. S. Daw, Phys. Rev. B 47, 10895 (1993).
- [13] T. Ozaki and K. Terakura, Phys. Rev. B 64, 195126 (2001).
- [14] S. Goedecker, Rev. of Mod. Phys. 71, 1085 (1999) and references therein.
- [15] J. M. Perez-Jorda, J. Chem. Phys. 101, 1738 (1994).
- [16] P. M. W. Gill, B. G. Johnson, and J. A. Pople, Chem. Phys. Lett. 209, 506 (1993).
- [17] C. Satoko, Phys. Rev. B 30, 1754 (1984).
- [18] A. D. Becke, J. Chem. Phys. 88, 2547 (1988).
- [19] M. R. Pederson and K. A. Jackson, Phys. Rev. B 41, 7453 (1990); K. Jackson and M. R. Pederson, Phys. Rev. B 42, 3276 (1990); A. Briley, M. R. Pederson, K. A. Jackson, D. C. Patton, and D. V. Porezag, Phys. Rev. B 58, 1786 (1998); D. Porezag, M. R. Pederson, and A. Y. Liu, Phys. Rev. B 60, 14132 (1999).
- [20] B. I. Dunlap and N. Rosch, J. de chimie physique 86, 671 (1989).
- [21] T. Ono and K. Hirose, Phys. Rev. Lett. 82, 5016 (1999).
- [22] J. R. Chelikowsky, N. Troullier, and Y. Saad, Phys. Rev. Lett. 72, 1240 (1994); J. R. Chelikowsky, X. Jing, K. Wu, and Y. Saad, Phys. Rev. B 53, 12071 (1996) and and references therein.
- [23] D. M. Ceperley and B. J. Alder, Phys. Rev. Lett. 45, 566 (1980); J. P. Perdew and A. Zunger, Phys. Rev. B 23, 5048 (1981).
- [24] J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996).
- [25] L. Kleinman and D. M. Bylander, Phys. Rev. Lett. 48, 1425 (1982).
- [26] P. E. Blochl, Phys. Rev. B 41, R5414 (1990).
- [27] W. H. Press, B. P. Flannery, S. A. Teukolsky, and W. T. Vetterling, Numerical Recipes (Cambridge University Press, Cambridge, 2002).
- [28] Our DFT code, OpenMX, the basis orbitals, and pseudo potentials used in this study are available on a web site (http://www.openmx-square.org/) in the constitution of the GNU General Public Licence.
- [29] S. A. Clough, Y. Beers, G. P. Klein, and L. S. Rothman, J. Chem. Phys. 59, 2254 (1973).
- [30] W. S. Benedict, N. Gailar, and E. K. Plyler, J. Chem. Phys. 24, 1139 (1956).
- [31] F. Gygi, Europhys. Lett. 19, 617 (1992); F. Gygi, Phys. Rev. B 48, 11692 (1993).