Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
ESM.direction x # x|y|z, default=x
The default direction is the x-axis.
Related information can be found in the
Effective screening medium method.
scf.eigen.lib elpa1 # elpa1|elpa2, default=elpa1
The default choice is ELPA1. Our benchmark calculations suggest that ELPA1 and ELPA2 are comparable
to each other with respect to the computational speed.
Related information can be found in the
ELPA|
ELPA.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
Thank you very much for your cooperation in advance.
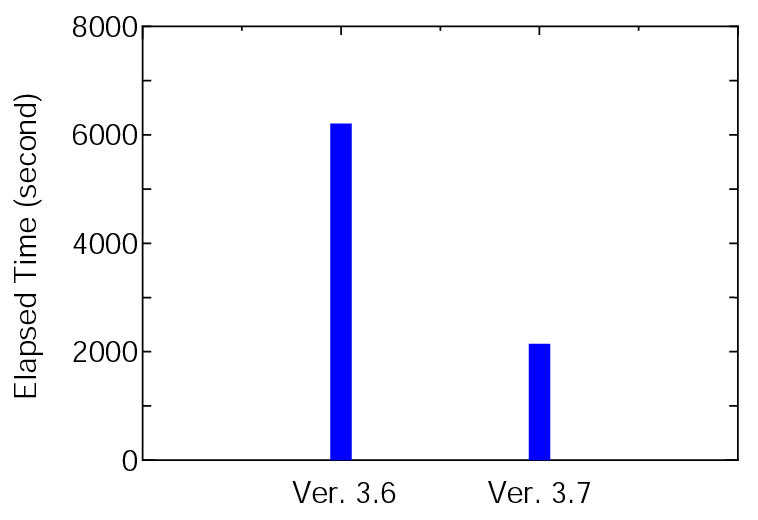
where 128 MPI processes and 2 OpenMP threads on CRAY-XC30 were used for both the versions. It is found that Ver. 3.7 is about three times faster than Ver. 3.6 for the benchmark sets. The elapsed time for each input file can be confirmed from 'runtestL.result_xc30_v3.6' and 'runtestL.result_xc30' stored in 'work/large_example'.
% mpirun -np 128 openmx -runtestL2 -nt 4
Then, OpenMX will run with 7 test files, and compare calculated results with the reference
results which are stored in 'work/large2_example'.
The following is a result of 'runtestL2' performed using 128 MPI processes and 4 OpenMP threads on CRAY-XC30.
| 1 | large2_example/C1000.dat | Elapsed time(s)= 1731.83 | diff Utot= 0.000000002838 | diff Force= 0.000000007504 |
| 2 | large2_example/Fe1000.dat | Elapsed time(s)=21731.24 | diff Utot= 0.000000010856 | diff Force= 0.000000000580 |
| 3 | large2_example/GRA1024.dat | Elapsed time(s)= 2245.67 | diff Utot= 0.000000002291 | diff Force= 0.000000015333 |
| 4 | large2_example/Ih-Ice1200.dat | Elapsed time(s)= 952.84 | diff Utot= 0.000000000031 | diff Force= 0.000000000213 |
| 5 | large2_example/Pt500.dat | Elapsed time(s)= 6831.16 | diff Utot= 0.000000002285 | diff Force= 0.000000004010 |
| 6 | large2_example/R-TiO2-1050.dat | Elapsed time(s)= 2259.97 | diff Utot= 0.000000000106 | diff Force= 0.000000001249 |
| 7 | large2_example/Si1000.dat | Elapsed time(s)= 1655.25 | diff Utot= 0.000000001615 | diff Force= 0.000000005764 |
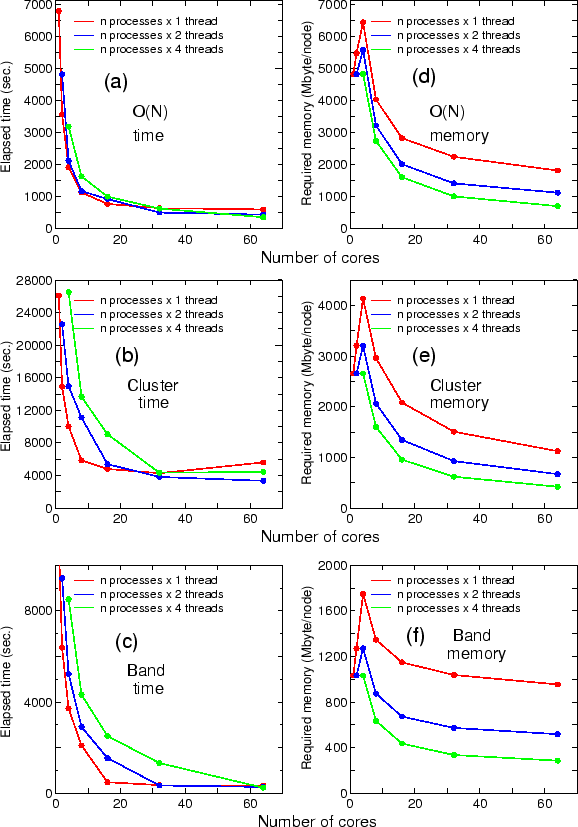
The quality of all the calculations is at a level of production run where double valence plus a single polarization functions are allocated to each atom as basis functions. Except for 'Pt500.dat', all the systems include more than 1000 atoms, where the last number of the file name implies the number of atoms for each system, and the elapsed time implies that geometry optimization for systems consisting of 1000 atoms is possible if several hundred processor cores are available. It is noted that the parallel eigenvalue solver introduced in OpenMX Ver. 3.7 is not exactly the same as ELPA1 distributed in here. Since we found that the original ELPA1 tends to encounter a numerical instability on some platforms, we modified ELPA1 to make it stabilized. The original parallel eigenvalue solver used in OpenMX Ver. 3.6 is also available by the following keyword:
scf.eigen.lib elpa1 # elpa1|lapack, default=elpa1
One can choose either 'elpa1' or 'lapack' depending on computational environment.
The default is 'elpa1'.
Due to the introduction of the ELPA based eigenvalue solver, users are requested to specify a FORTRAN compiler.
More information can be found in here and here.
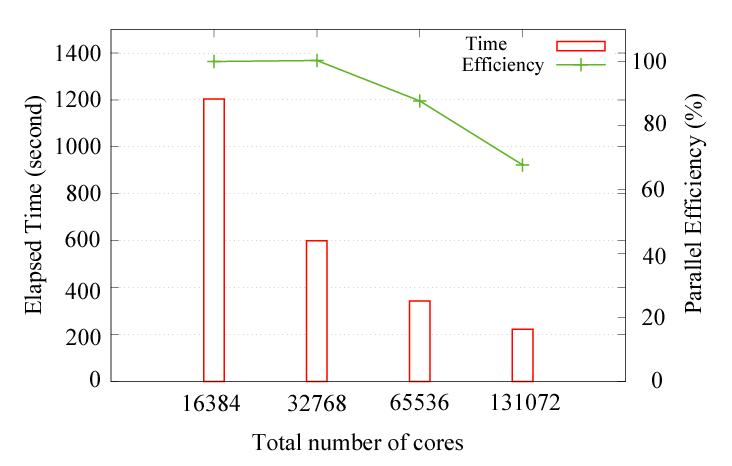
where the diamond structure consisting of 131072 carbon atoms was considered as a benchmark system, and eight OpenMP threads were used for all the cases. The parallel efficiency is about 68 % using 131072 cores by taking the case with 16384 cores as a reference. No additional keyword is introduced for the development. The new development has been already applied to two large-scale systems, resulting in two papers: J. Chem. Phys. 136, 134101 (2012) and Modelling Simul. Mater. Sci. Eng. 21, 045012 (2013).
Related information can be found in the manual.
MD.NEB.Parallel.Number 3
In this example, the calculations of every three images are parallelized at once where the MPI processes
are classified to three groups and utilized for the parallelization of each image among the three images.
In order to complete the calculations of all the images, the grouped calculations are repeated
by floor[(the number of images)/(MD.NEB.Parallel.Number)] times. The scheme may be useful for the NEB
calculation of a large-scale system. If the keyword is not specified in your input file, the default
parallelization scheme is employed.
Related information can be found in the
manual.
#pragma optimization_level 1Then, the optimization level is always set to 1 for those routines without depending on the optimization level user defines. The other purposes of the patch can be found at README.txt.
vps.type MBK
The MBK pseudopotential is a norm-conserving version of the Vanderbilt's ultrasoft
pseudopotential (Phys. Rev. B 41, 7892 (1990)). The feature allows us to take multiple
states with the same angular momentum quantum number into account for construction of
a separable pseudopotential. Thus, it is guranteed that the MBK scheme is more accurate
than the other norm-conserving schemes when semi-core states are included in the
construction of pseudopotential.
% mpirun -np 32 openmx DIA512-1.dat -nt 4 > dia512-1.std &
where '-nt' means the number of threads in each process managed by MPI.
If '-nt' is not specified, then the number of threads is set 1, which
corresponds to the pure MPI parallelization. For the installation,
please see the manual.
 |
AtomSpecies 6.2
total.electron 6.2
valence.electron 4.2
<ocupied.electrons
1 2.0
2 2.0 2.2
ocupied.electrons>
The above example is for a virtual atom on the way of carbon and
nitrogen atoms.
Also, it is noted that basis functions for the pseudopotential
of the virtual atom must be generated for the virtual atom with
the same fractional nuclear charge, since the atomic charge density
stored in *.pao is used to make the neutral atom potential.
As an illustration, the DOS of C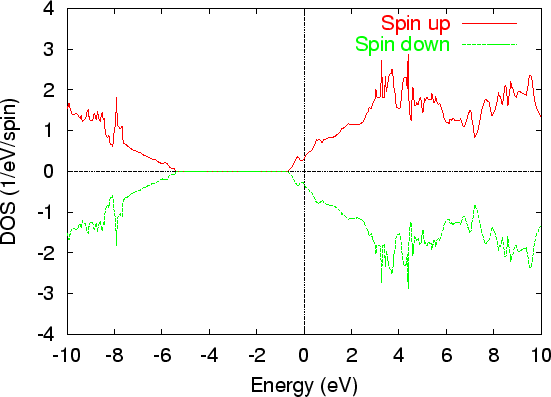 |
For the serial running
% ./openmx -runtestL
For the MPI parallel running
% mpirun -np 4 openmx -runtestL
For the OpenMP/MPI parallel running
% mpirun -np 4 openmx -runtestL -nt 1
Then, OpenMX will run with 20 test files, and compare calculated
results with the reference results which are stored in 'work/large_example'.
The comparison (absolute difference in the total energy and force) is
stored in a file 'runtestL.result' in the directory 'work'.
Since the automatic running test requires considerable memory size,
you may encounter a segmentation fault on computational
environment with small memory.
Also it is noted that the total elapsed time is more than
1 day even using 40 cores.
AtomSpecies 6.2
total.electron 6.2
valence.electron 4.2
<ocupied.electrons
1 2.0
2 2.0 2.2
ocupied.electrons>
The above example is for a virtual atom on the way of carbon and
nitrogen atoms. By just controling the above keywords,
you can easily generate pseudopotentials and basis functions
for virtual atoms. When you use those in OpenMX as input data,
no specification by keywords is required.
Please make sure that only OpenMX Ver. 3.4 or later accepts
the pseudopotentials and the basis functions for the virtual atoms.
Also, it is noted that basis functions for the pseudopotential
of the virtual atom must be generated for the virtual atom with
the same fractional nuclear charge, since the atomic charge density
stored in *.pao is used to make the neutral atom potential in OpenMX.
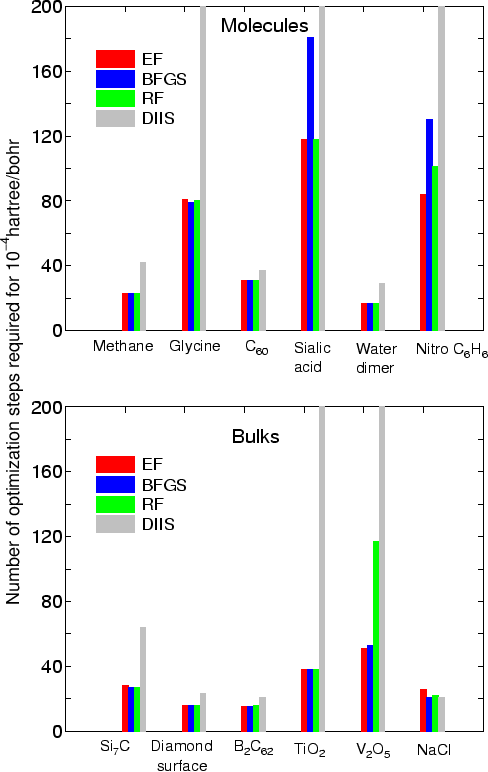
MD.Type EF # Opt|DIIS|BFGS|RF|EF MD.Opt.DIIS.History 3 # default=3 MD.Opt.StartDIIS 5 # default=5 MD.Opt.EveryDIIS 200 # default=200 MD.maxIter 100 # default=1 MD.Opt.criterion 1.0e-4 # default=0.0003 (Hartree/bohr)Since the usages of these keywords are same as in the previous version 3.2, you can find the details in the manual of the previous version 3.2. The initial step in the optimization is automatically tuned by monitoring the maximum force in the initial structure, while it was specified by the keyword "MD.Initial.MaxStep" in the previous version 3.2 (the keyword "MD.Initial.MaxStep" is not available in OpenMX3.3). As shown in the figure which shows the number of geometry steps to achieve the maximum force of below 0.0001 hartree/bohr in molecules and bulks, in most cases the EF method seems to be the most robust and efficient scheme, while the RF method also shows a good performance.
typedef float Type_DS_VNA; /* type of DS_VNA */ #define MPI_Type_DS_VNA MPI_FLOAT /* type of DS_VNA */ typedef float Type_Orbs_Grid; /* type of Orbs_Grid */ #define MPI_Type_Orbs_Grid MPI_FLOAT /* type of Orbs_Grid */By changing 'float' and 'MPI_FLOAT' into 'double' and 'MPI_DOUBLE', it is possible to allocate them in double-precision floating point if you like it.
<SO.factor
0 1.0
1 0.5
2 2.0
SO.factor>
The beginning of the description must be <SO.factor, and the last of
the description must be SO.factor>.
The number in the first column corresponds to that in the keyword 'pseudo.NandL',
and a scaling factor is given for each pseudopotential by the second column,
where '1.0' corresponds to the spin-orbit coupling in a real atom.
One can control the strength of spin-orbit coupling by changing the scaling factor.
Grid_Origin xxx yyy zzz
The values will be used in the following calculations (ii) and (iii).
scf.fixed.grid xxx yyy zzz
where 'xxx yyy zzz' is the coordinate of the origin you got in the calculation (i).
Then, you will have a cube file for charge (spin) density.
Let it 'A.cube'.
scf.fixed.grid xxx yyy zzz
where 'xxx yyy zzz' is the coordinate of the origin you got in the calculation (i).
Then, you will have a cube file for charge (spin) density.
Let it 'B.cube'.
% gcc diff_gcube.c -lm -o diff_gcube
% gcc add_gcube.c -lm -o add_gcube
(v) generate a cube file for differece charge (spin) density
% ./add_gcube A.cube B.cube A_B.cube
The file 'A_B.cube' is the cube file for the superposition of charge (spin) density
of two isolated systems.
Then, you can generate a cube file for the difference charge (spin) density induced
by the interaction as follows:
% ./diff_gcube AB.cube A_B.cube dAB.cube
The file 'dAB.cube' is the cube file for the difference charge (spin) density induced
by the interaction, where the difference means (AB - A_B).
DATA.PATH ../DFT_DATA2006/ # default=../DFT_DATA/
Both the absolute and relative specifications are available.
DosGauss.fileout on # default=off, on|off
DosGauss.Num.Mesh 200 # default=200
DosGauss.Width 0.2 # default=0.2 (eV)
When you use the scheme, specify 'on' for the keyword 'DosGauss.fileout'.
And the keyword 'DosGauss.Num.Mesh' gives the number of partitioning for the
energy range specified by the keyword 'Dos.Erange'. The keyword 'DosGauss.Width'
gives the width, a, of the Gaussian exp(-(E/a)^2). The keyword 'DosGauss.fileout'
and the keyword 'Dos.fileout' are mutually exclusive.
Therefore, when you use the scheme the keyword, 'Dos.fileout' must be 'off'
as follows:
Dos.fileout off # on|off, default=off
Also, the following two keywords are valid for both the keywords 'Dos.fileout' and
'DosGauss.file'.
Dos.Erange -20.0 20.0 # default=-20 20
Dos.Kgrid 5 5 5 # default=Kgrid1 Kgrid2 Kgrid3
It should be noted that the keyword 'DosGauss.fileout' generates only the Gaussian
broadening DOS, which means that the DOS by the tetrahedron method cannot be calculated
by the keyword 'DosGauss.fileout'.
scf.NC.Zeeman.Spin on # on|off, default=off
scf.NC.Mag.Field.Spin 1.0e+3 # default=0.0(Tesla)
When you include the Zeeman term for spin magnetic moment, switch on
the keyword 'scf.NC.Zeeman.Spin'.
The magnitude of the uniform magnetic field can be specified by the keyword
'scf.NC.Mag.Field.Spin' in units of Tesla. Moreover, we extend the scheme
as a constraint scheme in which the direction of the magnetic field can be
different from each atomic site atom by atom. Then, the direction of magnetic
field for spin magnetic moment can be controlled, for example, by the keyword
'Atoms.SpeciesAndCoordinates':
<Atoms.SpeciesAndCoordinates 1 Sc 0.000 0.000 0.000 6.6 4.4 10.0 50.0 160.0 20.0 1 on 2 Sc 2.000 0.000 0.000 6.6 4.4 80.0 50.0 160.0 20.0 1 on Atoms.SpeciesAndCoordinates>The 8th and 9th columns give the Euler angles, theta and phi, in order to specify the magnetic field for spin magnetic moment. The 12th column is a switch to the constraint. '1' means that the constraint is applied, and '0' no constraint. Since for each atomic site a different direction of the magnetic field can be applied, this scheme provides a way of studying non-collinear spin configuration. It is noted that the keyword 'scf.NC.Zeeman.Spin' and the keyword 'scf.Constraint.NC.Spin' are mutually exclusive. Therefore, when 'scf.NC.Zeeman.Spin' is 'on', the keyword 'scf.Constraint.NC.Spin' must be switched off as follows:
scf.Constraint.NC.Spin off # on|off, default=off
(6) Zeeman term for orbital magnetic moment
scf.NC.Zeeman.Orbital on # on|off, default=off
scf.NC.Mag.Field.Orbital 1.0e+3 # default=0.0(Tesla)
When you include the Zeeman term for orbital magnetic moment, switch on
the keyword 'scf.NC.Zeeman.Orbital'.
The magnitude of the uniform magnetic field can be specified by the keyword
'scf.NC.Mag.Field.Orbital' in units of Tesla. Moreover, we extend the scheme
as a constraint scheme in which the direction of the magnetic field can be
different from each atomic site atom by atom. Then, the direction of magnetic
field for orbital magnetic moment can be controlled, for example, by the keyword
'Atoms.SpeciesAndCoordinates':
<Atoms.SpeciesAndCoordinates 1 Sc 0.000 0.000 0.000 6.6 4.4 10.0 50.0 160.0 20.0 1 on 2 Sc 2.000 0.000 0.000 6.6 4.4 80.0 50.0 160.0 20.0 1 on Atoms.SpeciesAndCoordinates>The 10th and 11th columns give the Euler angles, theta and phi, in order to specify the magnetic field for orbital magnetic moment. The 12th column is a switch to the constraint. '1' means that the constraint is applied, and '0' no constraint. Since for each atomic site a different direction of the magnetic field can be applied, this scheme provides a way of studying non-collinear orbital configuration. Also, it is noted that the direction of magnetic field for orbital magnetic moment can be different from that for spin moment.
<Atoms.SpeciesAndCoordinates 1 Sc 0.000 0.000 0.000 6.6 4.4 10.0 50.0 160.0 20.0 1 on 2 Sc 2.000 0.000 0.000 6.6 4.4 80.0 50.0 160.0 20.0 1 on Atoms.SpeciesAndCoordinates>The specification of each column is listed as:
1: sequential serial number
2: species name
3: x-coordinate
4: y-coordinate
5: z-coordinate
6: initial occupation for up spin
7: initial occupation for down spin
8: Euler angle, theta, of the magnetic field for spin magnetic moment
9: Euler angle, phi, of the magnetic field for spin magnetic moment
Also, the 8th and 9th are used to generate the initial non-collinear
spin charge distribution
10: the Euler angle, theta, of the magnetic field for orbital magnetic moment
11: the Euler angle, phi, of the magnetic field for orbital magnetic moment
12: switch for the constraint schemes specified by the keywords
'scf.Constraint.NC.Spin', 'scf.NC.Zeeman.Orbital' and 'scf.NC.Zeeman.Orbital'.
'1' means that the constraint is applied, and '0' no constraint.
13: switch for enhancement of orbital polarization in the LDA+U method,
'on' means that the enhancement is made, 'off' no enhancement.
(8) Constrained geometry optimization and molecular dynamics
<MD.Fixed.XYZ
1 1 1 1
2 1 0 0
MD.Fixed.XYZ>
The example is for a system consisting of two atoms. If you have N atoms, then
you have to provide N-th rows in this specification. The 1st column is the same
sequential number to spefify atom as in the specification of the keyword
'Atoms.SpeciesAndCoordinates'. The 2nd, 3rd, 4th columns are switchs for
the x-, y-, z-coordinates. '1' means that the coordinate is fixed, and '0' relaxed.
It should be noted that the definition of the switch is
opposite
compared to the previous constraint schemes.
In above example, the x-, y-, z-coordinates of the atom '1' are fixed, only the
x-coordinate of the atom '2' is fixed. The default setting is that all the coordinates
are relaxed.
The fixing of atomic positions are valid
all the geometry optimizer and molecular dynamics schemes. So, the previous
constraint schemes such as 'Constraint_DIIS' are replaced by a combination of the keyword
'MD.Fixed.XYZ' and the geometry optimizer and molecular dynamics schemes specified
by the keyword 'MD.Type'.
Then, the following schemes are now available for the keyword 'MD.Type'.
NoMD
Opt
DIIS
NVE
NVT_VS
NVT_NH
(9) Initial velocity for molecular dynamics
<MD.Init.Velocity
1 3000.000 0.0 0.0
2 -3000.000 0.0 0.0
MD.Init.Velocity>
The example is for a system consisting of two atoms. If you have N atoms, then
you have to provide N-th rows in this specification. The 1st column is the same
sequential number to spefify atom as in the specification of the keyword
'Atoms.SpeciesAndCoordinates'. The 2nd, 3rd, and 4th columns are x-, y-,
and z-components of the velocity of each atom. The unit of the velocity is m/s.
The keyword 'MD.Init.Velocity' is compatible with the keyword 'MD.Fixed.XYZ'.
MD.Opt.DIIS.History 4 # default=4
MD.Opt.StartDIIS 5 # default=5
The keyword 'MD.Opt.DIIS.History' specifies the number of the previous
steps to estimate an optimum structure which will give the minimum norm
of forces used. The default value is 4.
Also, the geometry optimization step which starts 'DIIS' is specified
by the keyword 'MD.Opt.StartDIIS'. The geometry optimization steps
before starting DIIS type methods is performed by the steepest decent
method as in 'Opt'. The default value is 5.
For the details, see 'Section 13 Geometry optimization' in the manual.
scf.Mixing.EveryPulay # default = 5
The residual vectors in the Pulay-type mixing schemes tend to become
linearly dependent each other as the mixing steps accumulate, and
the linear dependence among the residual vectors makes the convergence
difficult. A way of avoiding the linear dependence is to do the Pulay-type
mixing occasionally during the Kerker mixing.
With this prescription, you can specify the frequency using the
keyword 'scf.Mixing.EveryPulay'.
For example, in case of 'scf.Mixing.EveryPulay=5', the Pulay-mixing is
made at every five SCF iteration, while Kerker-type mixing is used
at the other steps. 'scf.Mixing.EveryPulay=1' corresponds to the
conventional Pulay-type mixing. It is noted that the keyword
'scf.Mixing.EveryPulay' is supported for only 'RMM-DIISK', and
the default value is five.
For the details, see 'Section 11 SCF convergence' in the manual.
scf.Hubbard.U on # On|Off, default=off
For the details, see 'Section 29 LDA+U' in the manual.
scf.Constraint.NC.Spin on # on|off, default=off
scf.Constraint.NC.Spin.v 0.2 # default=0.0(eV)
You can switch on the keyword 'scf.Constraint.NC.Spin' and give
a magnitude by 'scf.Constraint.NC.Spin.v' which determines the
strength of constraint, when the constraint for the spin orientation
is introduced.
For the details, see 'Section 30 Constraint DFT for
non-collinear spin orientation' in the manual.
scf.lapack.dste dstegr # dstegr|dstedc|dstevx, default=dstegr
These lapack routines, dstegr, dstedc, and dstevx, are based on
a multiple relatively robust representation (MR3) scheme
(I.S.Dhillon and B.N.Parlett, SIAM J.Matrix Anal.Appl. 25, 858 (2004)),
a divide and conquer (DC) algorithm
(J.J.M.Cuppen, Numer.Math. 36, 177 (1981);
M.Gu and S.C.Eisenstat, SIAM J.Mat.Anal.Appl. 16, 172 (1995)),
and QR and inverse
iteration algorithm, respectively. For further details, see
the lapack website .
Based on our experiences, we find that the computational speed
is as follows:
dstevx < dstedc < dstegr
In contrast to the computational speed, the computational robustness
seems to be opposite as follows:
dstegr < dstedc < dstevx
So, an appropriate one (robuster and faster) on your computational
environment should be selected by this keyword, scf.lapack.dste.
scf.EigenvalueSolver GDC # DC|GDC|Cluster|Band
By optimizing the shell size in the divide-conquer (DC) method
(W.Yang, Phys.Rev.Lett. 66, 1438 (1991)), a generalized DC method
has been developed. The details will be published elsewhere.
scf.Mixing.Type Rmm-diisk # Simple|Rmm-Diis|GR-Pulay|Kerker|Rmm-Diisk
So, five mixing schemes are available in OpenMX2.3.
Although a mixing is performed for density matrices (real space)
in previous three mixing schemes, Simple, Rmm-Diis, and GR-Pulay,
the charge mixing is made in Fourier space in these newly
supported schemes, Kerker and Rmm-Diisk. So, the charge sloshing,
which comes from charge components with long wave length, can be
significantly suppressed by introducing Kerker's metric defined by
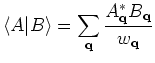
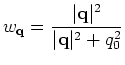
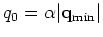
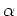 .
Then, the following keyword is available.
.
Then, the following keyword is available.
scf.Kerker.factor 1.0 # default=1.0
A larger significantly suppresses
the charge sloshing, but leads to slower convergence.
Since an optimum value depends on system, you may tune
an appropriate value for your system.
In addition, it should be noted that the relation between
'Kerker' and 'Rmm-Diisk' schemes is the same as that between
'Simple' and 'Rmm-Diis'.
Since all the keywords for 'Simple' and 'Rmm-Diis' are valid for
'Kerker' and 'Rmm-Diisk' as well, you may tune these parameters
to obtain the faster convergence for your systems.
LIB = -L/usr/local/lib -fftw3
If you want to use FFTW2, you need to add '-Dfftw2' for the compile option
as follows:
CC = gcc -Dfftw2
Since the computational time for FFT is a small fraction in the
total computational time, you can use either FFTW2 or FFTW3
without loosing significant efficiency.
For serial running
% ./openmx -runtest
For parallel running
% ./openmx -runtest "mpirun -np 4 openmx"
Then, OpenMX will run with several test files, and compare
calculated results with the previous results which are stored
in work/input_example. The comparison (difference in the total
energy and force) is stored in a file 'runtest.result'.
If the difference is within last seven digits, we may consider
that the installation is successful.
If you want to make reference files by youself, please execute
OpeMX as follows:
% ./openmx -maketest
Then, for *.dat files In work/input_example, OpenMX will generate
*.out files in work/input_example. So, you can add a new dat file
which is used in the next running test. But, please make sure
that the previous out files in work/input_example will be overwritten.
level.of.fileout 0 # default=1 (0-2)
Then, any Gaussian cube or grid file is not generated.
MD.Type NVT_VS # NOMD|Opt|NVE|NVT_VS|NVT_NH
Then, in this NVT molecular dynamics
the temperature for nuclear motion can be controlled by
<MD.TempControl
3
100 2 1000.0 0.0
400 10 700.0 0.4
700 40 500.0 0.7
MD.TempControl>
The beginning of the description must be <MD.TempControl, and
the last of the description must be MD.TempControl>.
The first number '3' gives the number of the following lines
to control the temperature. In this case you can see that
there are three lines. Following the number '3',
in the consecutive lines the first column
means the number of MD steps and the second column gives interval
of MD steps which determine ranges of MD steps and intervals
at which the velocity scaling is made.
For the above example, a velocity scaling is performed
at every two MD steps until 100 MD steps, at every 10 MD steps from 100
to 400 MD steps, and at every 40 MD steps from 400 to 700
MD steps. The third and fourth columns give a given temperature T_give
and a scaling parameter alpha in the interval.
In this velocity scaling velocities are scaled by
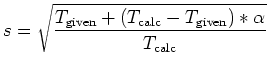
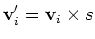
1: MD step
2: MD time
14: kinetic energy of nuclear motion, Ukc (Hartree)
15: DFT total energy, Utot (Hartree)
16: Utot + Ukc (Hartree)
17: Fermi energy (Hartree)
18: Given temperature for nuclear motion (K)
19: Calculated temperature for nuclear motion (K)
22: Nose-Hoover Hamitonian (Hartree)
which means that the first and second columns correspond to
MD step and MD time, and so on.
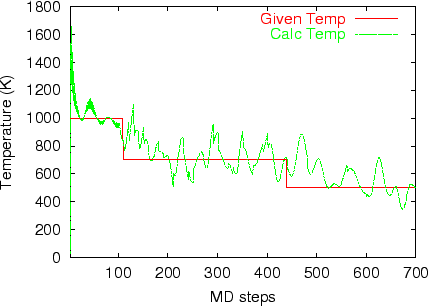
As an example, we show a result for the velocity scaling MD
of a glycine molecule
(input file)
in the following figure.
gnuplot> "gly_VS.ene" u 1:18 w l,"gly_VS.ene" u 1:19 w l
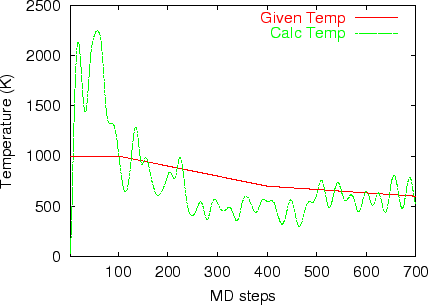
MD.Type NVT_NH # NOMD|Opt|NVE|NVT_VS|NVT_NH
Then, in this NVT molecular dynamics
the temperature for nuclear motion can be controlled by
<MD.TempControl
4
1 1000.0
100 1000.0
400 700.0
700 600.0
MD.TempControl>
The beginning of the description must be <MD.TempControl, and
the last of the description must be MD.TempControl>.
The first number '4' gives the number of the following lines
to control the temperature. In this case you can see that
there are four lines. Following the number '4',
in the consecutive lines the first and second columns
give the number of MD steps and a given temperature for
nuclear motion. The temperature between the interval is given
by a linear interpolation.
Although the same keyword 'MD.TempControl' as used in the
velocity scaling MD is utilized in this specification, it is noted
that the format is different from each other.
In addition to the specification of 'MD.TempControl', you must specify
a mass of heat bath by the following keyword:
NH.Mass.HeatBath 30.0 # default = 20.0
In this specification, we use a unit that the weight of a proton is 1.0.
Calculated quantities at every MD step are stored in an output
file '*.ene' as explained in 'Velocity scaling molecular dynamics'.
As an example, we show a result for Nose-Hoover MD
of a glycine molecule (input file)
in the following figure.
gnuplot> "gly_NH.ene" u 1:18 w l,"gly_NH.ene" u 1:19 w l
MD.Type NVE # NOMD|Opt|NVE|NVT_VS|NVT_NH
It should be noted that the old option 'Constant_Energy_MD'
is not available anymore.
scf.SpinOrbit.Coupling on # On|Off, default=off
If you use a fully relativistic j-dependent pseudo potential,
and set the keyword 'OFF', then the j-dependent pseudo potential
are automatically averaged with a weight of j-degeneracy when
it is read by OpenMX, which corresponds to a scalar relativistic
pseudo potential.
See also the Section 'Relativistic effects' in the manual
for the details.
scf.SpinPolarization NC # On|Off|NC
If the option 'NC' is specified, wave functions are expressed by
a two components spinor. An initial spin orientation of each site
is given by
<Atoms.SpeciesAndCoordinates
1 Cr 1.07400 1.07400 0.00000 4.0 8.0 90.0 90.0 1
2 Cr -1.07400 1.07400 0.00000 4.0 8.0 90.0 -90.0 1
3 Cr -1.07400 -1.07400 0.00000 4.0 8.0 90.0 90.0 1
4 Cr 1.07400 -1.07400 0.00000 4.0 8.0 90.0 -90.0 1
5 Cr 0.00000 0.00000 1.90000 7.0 5.0 0.0 0.0 1
Atoms.SpeciesAndCoordinates>
The Euler angles, theta and phi, to determine the spin
orientation are given by the 8th and 9th columns in the specifination
of '<Atoms.SpeciesAndCoordinates'. The final 10th column is a switch
for determining whether the spin orientation is relaxed during
the SCF calculation. '1' means that the spin orientation is relaxed.
'0' means that the spin orientation is fixed at the initial orientation.
See also the Section 'Non-collinear DFT' in the manual
for the details.
scf.system.charge 1.0 # default=0.0
The plus and minus signs correspond to hole and electron dopings,
respectively.
A partial charge doping is also possible. The excess charge
given by the keyword 'scf.system.charge' is compensated
by a uniform background opposite charge, since FFT is used
to solve Poisson's equation in OpenMX.
Voronoi.charge on # on|off, default = off
The result is stored in *.out.
scf.ProExpn.VNA on # on|off, default = on
If the keyword is 'on', this means the projector expansion, otherwise
the real space grid integration of the neutral atom.
eq.type dirac # sch|sdirac|dirac
where 'sch', 'sdirac', and 'dirac' mean the Schrodinger equation
(no relativistic effect), a scalar relativistic treatment, and
a full relativistic treatment of Dirac equation, respectively.
Although eq.type=dirac means the scalar relativistic treatment
in ADPACK1.3,
the scalar relativistic treatment is specified by 'sdirac' in ADPACK1.5.
In the scalar relativistic treatment, the coupled Dirac equations
are averaged with a weight of j-degeneracy, and solved
by taking account of both the majority and minority components of
radial wave function. Thus, the scalar relativistic treatment
includes explicitly kinematic relativistic effects (Darwin and
mass velocity terms), and implicitly averaged
spin-orbit coupling (no energy splitting).
On the other hand, in the full relativistic treatment,
j-dependent Dirac equations are solved including both the majority
and minority components of radial wave function.
Thus, energy splitting by spin-orbit coupling is also considered.
System.UseRestartfile YES # NO|YES, default=NO
System.Restartfile C0_Rest # default=null
If the keyword, System.UseRestartfile,
is specified as YES, a restart file which contains informations of all
electron calculation is used in order to skip all electron calculation.
If there is no restart file, a restart file is generated in case of
System.UseRestartfile=YES.
If System.UseRestartfile=YES, then the name specified by the keyword,
System.Restartfile, is refered to as a restart file.
scf.XcType GGA-PBE
You can specify either the non-spin or spin polarization calculation
based on GGA using the keyword, scf.SpinPolarization.
scf.Electric.Field 1.0 0.0 0.0
The sign of electric field is for electrons.
When the uniform external electric field is applied to
a periodic system, discontinuities of the potential are
introduced. For molecular systems, the discontinuities are located
in the vacuum resion. Thus, numerical instabilities may not be induced.
On the other hand, you might meet numerical instabilities due to the
discontinuities for bulk systems.
orbitalOpt.Method species
The PAOs for proteins, DNA, and RNA were optimized
using the constrained orbital optimization scheme.
<Atoms.SpeciesAndCoordinates
1 C 0.300000 0.000000 0.000000 2.0 2.0 0
2 H -0.889981 -0.629312 0.000000 0.5 0.5 0
3 H 0.000000 0.629312 -0.889981 0.5 0.5 1
4 H 0.000000 0.629312 0.889981 0.5 0.5 1
5 H 0.889981 -0.629312 0.000000 0.5 0.5 1
Atoms.SpeciesAndCoordinates>
In the final column, 0 means that the atom is fixed at the initial
position, and 1 means that the atom is relaxed. If you specify the other
keywords for MD.type, then you do not need add the number in the final
column.
<Atoms.SpeciesAndCoordinates
1 C 0.000000 0.000000 0.000000 2.0 2.0
2 H -0.889981 -0.629312 0.000000 0.5 0.5
3 H 0.000000 0.629312 -0.889981 0.5 0.5
4 H 0.000000 0.629312 0.889981 0.5 0.5
5 H 0.889981 -0.629312 0.000000 0.5 0.5
Atoms.SpeciesAndCoordinates>
Please don't forget to attach a sequential serial number
for identifying atoms in the first column.
rho_inp = rho_opt + alpha*R_opt,
where alpha is a parameter, ranging from 0 to 1,
which can vary automatically.
When 'scf.Mixing.Type=Rmm-Diis' or 'scf.Mixing.Type=Gr-Pulay' is
used, the following recipes are helpful to obtain the convergence
of SCF calculations: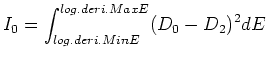
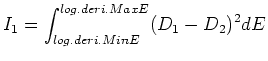
<log.deri.R
0 2.0
1 2.0
log.deri.R>
The beginning of the description must be <log.deri.R, and
the last of the description must be log.deri.R>.
The first column is the angular momentum number L, and the second
column is the radius at which the logarithmic derivatives of
radial wave functions are evaluated.
<pseudo.NandL
0 3 0 1.8
1 3 1 2.3
2 4 0 1.8
3 4 1 2.3
pseudo.NandL>