The DC method is a robust scheme and can be applicable to a wide variety
of materials with a reasonable degree of accuracy and efficiency, while
this scheme is suitable especially for covalent systems.
In this subsection, the O(![]() ) calculation using the DC method is
illustrated. In an input file 'DIA8_DC.dat' which can be found in the
directory 'work', please specify DC for
the keyword 'scf.EigenvalueSolver'.
) calculation using the DC method is
illustrated. In an input file 'DIA8_DC.dat' which can be found in the
directory 'work', please specify DC for
the keyword 'scf.EigenvalueSolver'.
scf.EigenvalueSolver DC
Then, one can execute OpenMX by:
% ./openmx DIA8_DC.dat
The input file is for an O(
The keyword 'orderN.HoppingRanges'
defines the radius of a sphere which is centered on each atom.
The physically truncated cluster for each atom is constructed by
picking up atoms inside the sphere with the radius in the DC and O(![]() )
Krylov subspace methods.
)
Krylov subspace methods.
If the number of atoms in the systems is N, N small eigenvalue problems for the N physically truncated clusters are solved, and then the total density of states (DOS) is constructed as the sum of the projected DOS of each physically truncated cluster. Although the appropriate value for 'orderN.HoppingRanges' depends on systems, for molecular systems the following values are recommended as a trade-off between the computational accuracy and efficiency:
orderN.HoppingRanges 6.0 - 7.0
Table 2 shows the comparison in the total energy between the exact
diagonalization and the DC method for a C![]() molecule and small
peptide molecules (valorphin [63]), and DNA consisting of
cytosines and guanines.
We find that errors in the total energy calculated by the DC method are
about a few mHartree in the system size.
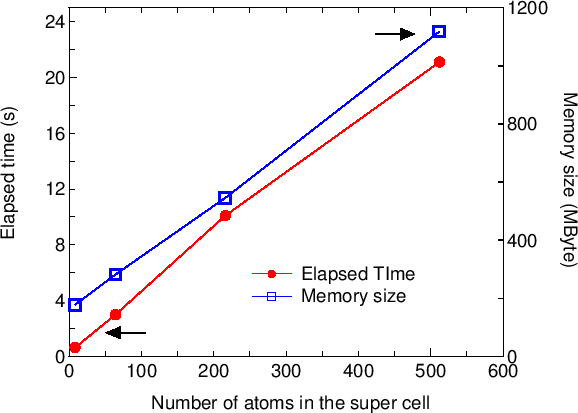
Also, it can be estimated that the DC method is faster
than the conventional diagonalization when the number of atoms is
larger than 500 atoms, while the crossing point between the conventional
diagonalization and the DC method with respect to computational time
depends on systems and the number of processors in the parallel calculation.
molecule and small
peptide molecules (valorphin [63]), and DNA consisting of
cytosines and guanines.
We find that errors in the total energy calculated by the DC method are
about a few mHartree in the system size.
Also, it can be estimated that the DC method is faster
than the conventional diagonalization when the number of atoms is
larger than 500 atoms, while the crossing point between the conventional
diagonalization and the DC method with respect to computational time
depends on systems and the number of processors in the parallel calculation.
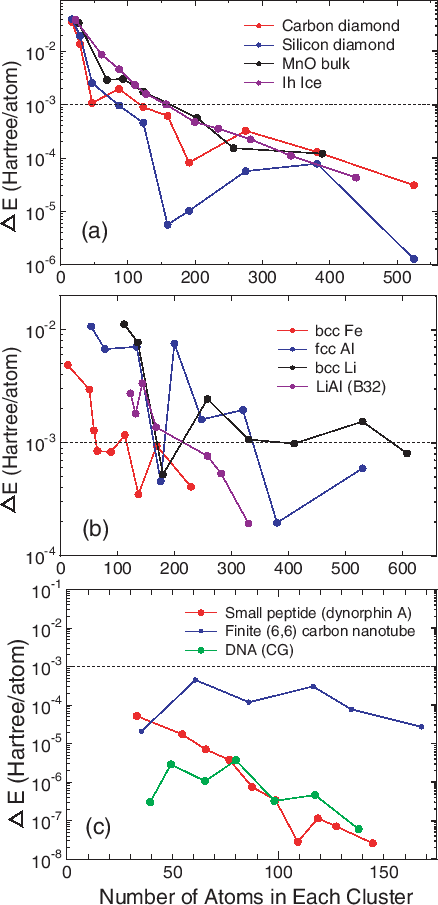
To see an overall tendency in the convergence properties of total energy with respect to the size of truncated cluster, the error in the total energy, compared to the exact diagonalization, is shown as a function of the number of atoms in each cluster for (a) bulks with a finite gap, (b) metals, and (c) molecular systems in Fig. 16. We see that the error decreases almost exponentially for the bulks with a finite gap and molecular systems, while the convergence speed is slower for metals.
|
 |