



Next: Scalar relativistic treatment
Up: Relativistic effects
Previous: Relativistic effects
Contents
Index
The fully relativistic effects including the spin-orbit coupling
within the pseudopotential scheme can be included in the non-collinear
DFT calculations [10,19,13],
while the inclusion of the spin-orbit coupling is not supported in the collinear
DFT calculation. The inclusion of fully relativistic effects is made
by the following two steps:
(1) Making of j-dependent pseudopotentials
First, you are requested to generate j-dependent pseudopotentials
using ADPACK. For your convenience, the j-dependent pseudopotentials
are available for many elements in the database Ver. 2013 [89].
The details how to make the j-dependent pseudopotential are found in
the manual of ADPACK.
(2) SCF calculation
If you specify j-dependent pseudopotentials in the specification of
' Definition.of.Atomic.Species',
it is possible to include spin-orbit
coupling by the following keyword 'scf.SpinOrbit.Coupling':
Definition.of.Atomic.Species',
it is possible to include spin-orbit
coupling by the following keyword 'scf.SpinOrbit.Coupling':
scf.SpinOrbit.Coupling on # On|Off, default=off
Then, the spin-orbit coupling can be self-consistently incorporated
within the pseudopotential scheme rather than a perturbation scheme.
Due to the spin-orbit coupling, 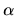 and
and 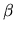 spin components
in the two component spinor can directly interact.
In order to determine the absolute spin orientation in the non-collinear
DFT calculations, you have to include the spin-orbit coupling, otherwise
the spin orientation is not uniquely determined in real space.
As an illustration of spin-orbit splitting, we show band structures of
a bulk GaAs calculated by the non-collinear DFT without and with spin-orbit
coupling in Fig. 26, where the input file is 'GaAs.dat' in the
directory 'work'.
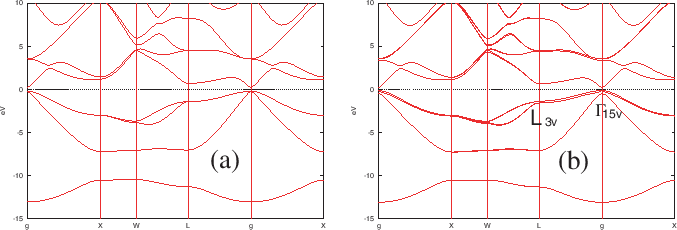
In Fig. 26(b) we can see that there are spin-orbit
splittings in the band dispersion, while no spin-orbit splitting is
observed in Fig. 26(a). The spin-orbit splittings at two k-points,
spin components
in the two component spinor can directly interact.
In order to determine the absolute spin orientation in the non-collinear
DFT calculations, you have to include the spin-orbit coupling, otherwise
the spin orientation is not uniquely determined in real space.
As an illustration of spin-orbit splitting, we show band structures of
a bulk GaAs calculated by the non-collinear DFT without and with spin-orbit
coupling in Fig. 26, where the input file is 'GaAs.dat' in the
directory 'work'.
In Fig. 26(b) we can see that there are spin-orbit
splittings in the band dispersion, while no spin-orbit splitting is
observed in Fig. 26(a). The spin-orbit splittings at two k-points,
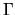 and
and 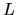 , are listed together with the other calculations
and experimental values in Table 3.
We see a good agreement in this table.
, are listed together with the other calculations
and experimental values in Table 3.
We see a good agreement in this table.
Figure 26:
Band structures of a bulk GaAs calculated by the non-collinear DFT
(a) without and (b) with the spin-orbit coupling.
In these calculations, Ga7.0-s2p2d2 and As7.0-s2p2d2 were used
as a basis set, and Ga_CA13.vps and As_CA13.vps were used for
pseudopotentials, which are stored in the database Ver. 2013.
For the exchange-correlation terms, LDA was used.
We used 12 12 12 and 140 (Ryd)
for scf.Kgrid and scf.energycutoff,
respectively.
Also the experimental value (5.65Å) was used for the lattice
constant.
The input file is 'GaAs.dat' in the directory 'work'.
12 12 and 140 (Ryd)
for scf.Kgrid and scf.energycutoff,
respectively.
Also the experimental value (5.65Å) was used for the lattice
constant.
The input file is 'GaAs.dat' in the directory 'work'.
 |
Next: Scalar relativistic treatment
Up: Relativistic effects
Previous: Relativistic effects
Contents
Index
t-ozaki
2013-05-22