Next: Exchange coupling parameter Up: User's manual of OpenMX Previous: Zeeman term for orbital Contents Index
The macroscopic electric polarization of a bulk system can
be calculated based on the Berry phase formalism [14].
As an example, let us illustrate a calculation of a Born
effective charge of Na in a NaCl bulk via the macroscopic
polarization.
(1) SCF calculation
First, perform a conventional SCF calculation using an input
file 'NaCl.dat' in the directory 'work'. Then, the following
keyword 'HS.fileout' should be switched on
HS.fileout on # on|off, default=off
When the calculation is completed normally, then you can find
an output file 'nacl.scfout' in the directory 'work'.
(2) Calculation of macroscopic polarization
The macroscopic polarization is calculated by a post-processing
code 'polB' of which input data is 'nacl.scfout'.
In the directory 'source', compile as follows:
% make polB
When the compile is completed normally, then you can find
an executable file 'polB' in the directory 'work'.
Then, move to the directory 'work', and perform as follows:
% polB nacl.scfout
or
% polB nacl.scfout < in > out
In the latter case, the file 'in' contains the following ingredients:
9 9 9
1 1 1
In the former case, you will be interactively asked from the program as follows:
****************************************************************** ****************************************************************** polB: code for calculating the electric polarization of bulk systems Copyright (C), 2006-2007, Fumiyuki Ishii and Taisuke Ozaki This is free software, and you are welcome to redistribute it under the constitution of the GNU-GPL. ****************************************************************** ****************************************************************** Read the scfout file (nacl.scfout) Previous eigenvalue solver = Band atomnum = 2 ChemP = -0.156250000000 (Hartree) E_Temp = 300.000000000000 (K) Total_SpinS = 0.000000000000 (K) Spin treatment = collinear spin-unpolarized r-space primitive vector (Bohr) tv1= 0.000000 5.319579 5.319579 tv2= 5.319579 0.000000 5.319579 tv3= 5.319579 5.319579 0.000000 k-space primitive vector (Bohr^-1) rtv1= -0.590572 0.590572 0.590572 rtv2= 0.590572 -0.590572 0.590572 rtv3= 0.590572 0.590572 -0.590572 Cell_Volume=301.065992 (Bohr^3) Specify the number of grids to discretize reciprocal a-, b-, and c-vectors (e.g 2 4 3) k1 0.00000 0.11111 0.22222 0.33333 0.44444 ... k2 0.00000 0.11111 0.22222 0.33333 0.44444 ... k3 0.00000 0.11111 0.22222 0.33333 0.44444 ... Specify the direction of polarization as reciprocal a-, b-, and c-vectors (e.g 0 0 1 ) 1 1 1Then, the calculation will start like this:
calculating the polarization along the a-axis ....
The number of strings for Berry phase : AB mesh=81
calculating the polarization along the a-axis .... 1/ 82
calculating the polarization along the a-axis .... 2/ 82
.....
...
*******************************************************
Electric dipole (Debye) : Berry phase
*******************************************************
Absolute dipole moment 163.93373639
Background Core Electron Total
Dx -0.00000000 94.64718996 -0.00000338 94.64718658
Dy -0.00000000 94.64718996 -0.00000283 94.64718713
Dz -0.00000000 94.64718996 -0.00000317 94.64718679
***************************************************************
Electric polarization (muC/cm^2) : Berry phase
***************************************************************
Background Core Electron Total
Px -0.00000000 707.66166752 -0.00002529 707.66164223
Py -0.00000000 707.66166752 -0.00002118 707.66164633
Pz -0.00000000 707.66166752 -0.00002371 707.66164381
Elapsed time = 77.772559 (s) for myid= 0
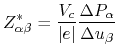 |
Px = 94.39497736 (Debye/unit cell) at x= -0.05 (Ang)
Px = 94.64718658 (Debye/unit cell) at x= 0.0 (Ang)
Px = 94.89939513 (Debye/unit cell) at x= 0.05 (Ang)
 |
|||
| OpenMX | FD | Expt. | |
| 1.05 | 1.09 | 1.12 |
Note that in the NaCl bulk the off-diagonal terms in the tensor of Born charge are
zero, and
![]() . In Table 6 we see that
the calculated value is in good agreement with the other calculation
[77] and an experimental result [78].
The calculation of macroscopic polarization is supported for both the
collinear and non-collinear DFT. It is also noted that the code 'polB'
has been parallelized for large-scale systems where the number of processors
can exceed the number of atoms in the system.
. In Table 6 we see that
the calculated value is in good agreement with the other calculation
[77] and an experimental result [78].
The calculation of macroscopic polarization is supported for both the
collinear and non-collinear DFT. It is also noted that the code 'polB'
has been parallelized for large-scale systems where the number of processors
can exceed the number of atoms in the system.