Next: Exchange coupling parameter Up: User's manual of OpenMX Previous: Zeeman term for orbital Contents Index
The macroscopic electric polarization of a bulk system can
be calculated based on the Berry phase formalism [15].
As an example, let us illustrate a calculation of a Born
effective charge of Na in a NaCl bulk via the macroscopic
polarization.
(1) SCF calculation
First, perform a conventional SCF calculation using an input
file 'NaCl.dat' in the directory 'work'. Then, the following
keyword 'HS.fileout' should be switched on
HS.fileout on # on|off, default=off
When the calculation is completed normally, then you can find
an output file 'nacl.scfout' in the directory 'work'.
(2) Calculation of macroscopic polarization
The macroscopic polarization is calculated by a post-processing
code 'polB' of which input data is 'nacl.scfout'.
In the directory 'source', please compile as follows:
% make polB
When the compilation is completed normally, then you can find
an executable file 'polB' in the directory 'work'.
Then, please move to the directory 'work', and perform as follows:
% polB nacl.scfout
or
% polB nacl.scfout < in > out
In the latter case, the file 'in' contains the following ingredients:
9 9 9
1 1 1
In the former case, you will be interactively asked from the program as follows:
****************************************************************** ****************************************************************** polB: code for calculating the electric polarization of bulk systems Copyright (C), 2006-2007, Fumiyuki Ishii and Taisuke Ozaki This is free software, and you are welcome to redistribute it under the constitution of the GNU-GPL. ****************************************************************** ****************************************************************** Read the scfout file (nacl.scfout) Previous eigenvalue solver = Band atomnum = 2 ChemP = -0.156250000000 (Hartree) E_Temp = 300.000000000000 (K) Total_SpinS = 0.000000000000 (K) Spin treatment = collinear spin-unpolarized r-space primitive vector (Bohr) tv1= 0.000000 5.319579 5.319579 tv2= 5.319579 0.000000 5.319579 tv3= 5.319579 5.319579 0.000000 k-space primitive vector (Bohr^-1) rtv1= -0.590572 0.590572 0.590572 rtv2= 0.590572 -0.590572 0.590572 rtv3= 0.590572 0.590572 -0.590572 Cell_Volume=301.065992 (Bohr^3) Specify the number of grids to discretize reciprocal a-, b-, and c-vectors (e.g 2 4 3) k1 0.00000 0.11111 0.22222 0.33333 0.44444 ... k2 0.00000 0.11111 0.22222 0.33333 0.44444 ... k3 0.00000 0.11111 0.22222 0.33333 0.44444 ... Specify the direction of polarization as reciprocal a-, b-, and c-vectors (e.g 0 0 1 ) 1 1 1Then, the calculation will start like this:
calculating the polarization along the a-axis ....
The number of strings for Berry phase : AB mesh=81
calculating the polarization along the a-axis .... 1/ 82
calculating the polarization along the a-axis .... 2/ 82
.....
...
*******************************************************
Electric dipole (Debye) : Berry phase
*******************************************************
Absolute dipole moment 163.93373639
Background Core Electron Total
Dx -0.00000000 94.64718996 -0.00000338 94.64718658
Dy -0.00000000 94.64718996 -0.00000283 94.64718713
Dz -0.00000000 94.64718996 -0.00000317 94.64718679
***************************************************************
Electric polarization (muC/cm^2) : Berry phase
***************************************************************
Background Core Electron Total
Px -0.00000000 707.66166752 -0.00002529 707.66164223
Py -0.00000000 707.66166752 -0.00002118 707.66164633
Pz -0.00000000 707.66166752 -0.00002371 707.66164381
Elapsed time = 77.772559 (s) for myid= 0
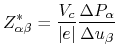 |
Px = 94.39497736 (Debye/unit cell) at x= -0.05 (Ang)
Px = 94.64718658 (Debye/unit cell) at x= 0.0 (Ang)
Px = 94.89939513 (Debye/unit cell) at x= 0.05 (Ang)
 |
|||
| OpenMX | FD | Expt. | |
| 1.05 | 1.09 | 1.12 |
Note that in the NaCl bulk the off-diagonal terms in the tensor of Born charge are
zero, and
![]() . In Table 7 we see that
the calculated value is in good agreement with the other calculation
[116] and an experimental result [117].
The calculation of macroscopic polarization is supported for both the
collinear and non-collinear DFT. It is also noted that the code 'polB'
has been parallelized for large-scale systems where the number of processors
can exceed the number of atoms in the system.
. In Table 7 we see that
the calculated value is in good agreement with the other calculation
[116] and an experimental result [117].
The calculation of macroscopic polarization is supported for both the
collinear and non-collinear DFT. It is also noted that the code 'polB'
has been parallelized for large-scale systems where the number of processors
can exceed the number of atoms in the system.