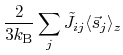Next: Electric transport calculations Up: Exchange coupling parameter Previous: Execution of jx and Contents Index
As an example of the cluster case, using an input file 'Fe_Cluster_jx.dat' which is stored in the directory 'work', you can first perform a conventional calculation using 'openmx' as
% mpirun -np 2 ./openmx Fe_Cluster_jx.dat
The input file 'Fe_Cluster_jx.dat' is for the SCF calculation of a iron dimer.
After finishing the calculation normally, you can obtain a scfout file 'Fe_Cluster_jx.scfout'.
Then the calculation by 'jx' is performed as
% ./jx Fe_Cluster_jx.scfout jx_cluster.config
where 'jx_cluster.config' is also available in the directory 'work'.
Then, you may see the following message on your screen.
********************************************************************
********************************************************************
jx: code for calculating an effective exchange coupling constant J
Copyright (C), 2003, Myung Joon Han, Jaejun Yu, and Taisuke Ozaki
2019, Asako Terasawa and Taisuke Ozaki
This is free software, and you are welcome to
redistribute it under the constitution of the GNU-GPL.
********************************************************************
********************************************************************
Read the scfout file (Fe_Cluster_jx.scfout)
***
The file format of the SCFOUT file: 3
And it supports the following functions:
- jx
- polB
- kSpin
- Z2FH
- calB
***
Previous eigenvalue solver = Cluster
atomnum = 2
ChemP = -0.089740215968 (Hartree)
E_Temp = 300.000000000000 (K)
Evaluation of J based on cluster calculation
i j J [meV] J [mRy]
-------------------------------------------------
1 1 1591.520791120630 116.974621661729
1 2 106.511867492210 7.828477938772
2 2 1591.520746009061 116.974618346089
Elapsed time = 0.036225 (s)
The exchange coupling constant As an example of the bulk case, you can perform a conventional calculation using 'openmx' and an input file 'Fe_Bulk_jx.dat' available in the directory 'work' as
% mpirun -np 28 ./openmx Fe_Bulk_jx.dat
The input file 'Fe_Bulk_jx.dat' is for the SCF calculation of a BCC iron.
After finishing the calculation normally, you can obtain a scfout file 'Fe_Bulk_jx.scfout'.
Then the calculation by 'jx' is performed as
% mpirun -np 112 ./jx Fe_Bulk_jx.scfout Fe_Bulk_jx.config | tee jx.log
where 'Fe_Bulk_jx.config' is available in the directory 'work'.
Then, you may see the following message on your screen.
********************************************************************
********************************************************************
jx: code for calculating an effective exchange coupling constant J
Copyright (C), 2003, Myung Joon Han, Jaejun Yu, and Taisuke Ozaki
2019, Asako Terasawa and Taisuke Ozaki
This is free software, and you are welcome to
redistribute it under the constitution of the GNU-GPL.
********************************************************************
********************************************************************
Read the scfout file (Fe_Bulk_jx.scfout)
***
The file format of the SCFOUT file: 3
And it supports the following functions:
- jx
- polB
- kSpin
- Z2FH
- calB
***
Previous eigenvalue solver = Band
atomnum = 2
ChemP = -0.205912787451 (Hartree)
E_Temp = 300.000000000000 (K)
Jij calculation for a periodic structure
Number of k-grids: 27 27 27
flag_periodic_sum = 0: coupling between site i at cell 0 and site j at cell R
Number of poles of Fermi-Dirac continued fraction (PRB.75.035123): 60
i j c1 c2 c3 J [meV] J [mRy] time_eig [s] ...
---------------------------------------------------------------------------------- ...
1 1 -2 -2 -2 -0.845809571401 -0.062165857439 0.51534 ...
1 1 -2 -2 -1 0.274300677331 0.020160728111 0.00000 ...
1 1 -2 -2 0 0.036006012552 0.002646393135 0.00000 ...
1 1 -2 -2 1 0.274300705154 0.020160730156 0.00000 ...
1 1 -2 -2 2 -0.845809596417 -0.062165859278 0.00000 ...
1 1 -2 -1 -2 0.274300737539 0.020160732536 0.00000 ...
1 1 -2 -1 -1 -0.206315672897 -0.015163922403 0.00000 ...
1 1 -2 -1 0 0.149714301525 0.011003798302 0.00000 ...
1 1 -2 -1 1 -0.206315540488 -0.015163912672 0.00000 ...
1 1 -2 -1 2 0.274300804604 0.020160737465 0.00000 ...
...
..
2 2 0 -1 2 0.149714016159 0.011003777328 0.00000 ...
2 2 0 0 -2 0.401809366424 0.029532443987 0.00000 ...
2 2 0 0 -1 11.452192349598 0.841720620155 0.00000 ...
Elapsed time = 340.817975 (s)
In Fig. 38 (a), the obtained exchange coupling constant
It is also possible to calculate the Curie temperature of periodic systems from calculated exchange coupling
constants and the mean field approximation.
That is, the Curie temperature
![]() of general periodic system can
be obtained as the maximum eigenvalue of the following eigenvalue equation:
of general periodic system can
be obtained as the maximum eigenvalue of the following eigenvalue equation:
![\includegraphics[width=160mm]{jx_examples.eps}](img268.png)
|
| |||||||||||||||